Li Ao, Liu Yuanning, Zhao Qihong, Feng Huanqing, Harris Lyndsay, Wang Minghui
Centers for Biomedical Engineering, University of Science and Technology of China, Hefei, China ; School of Information Science and Technology, University of Science and Technology of China, Hefei, China.
School of Information Science and Technology, University of Science and Technology of China, Hefei, China.
PLoS One. 2014 Jan 30;9(1):e87212. doi: 10.1371/journal.pone.0087212. eCollection 2014.
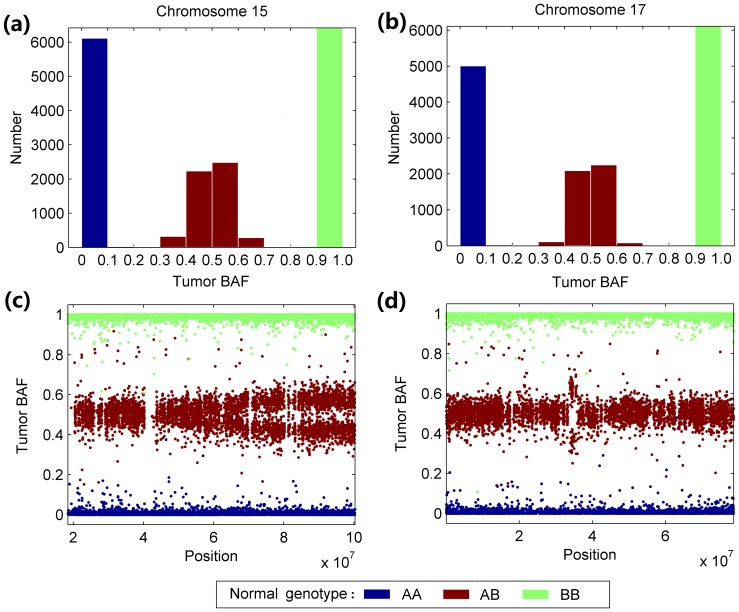
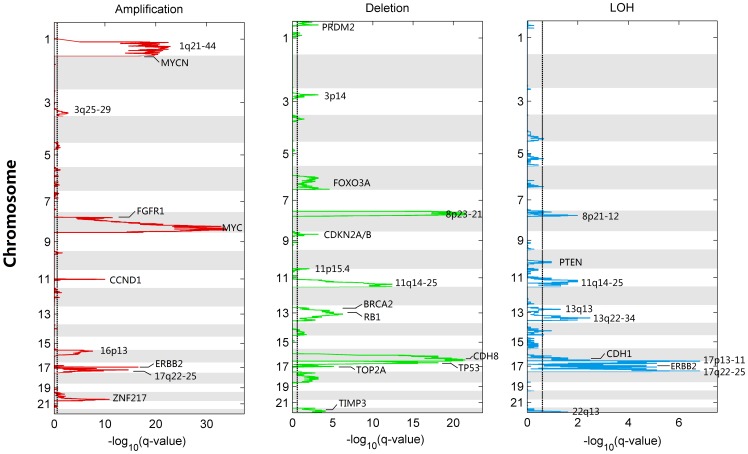
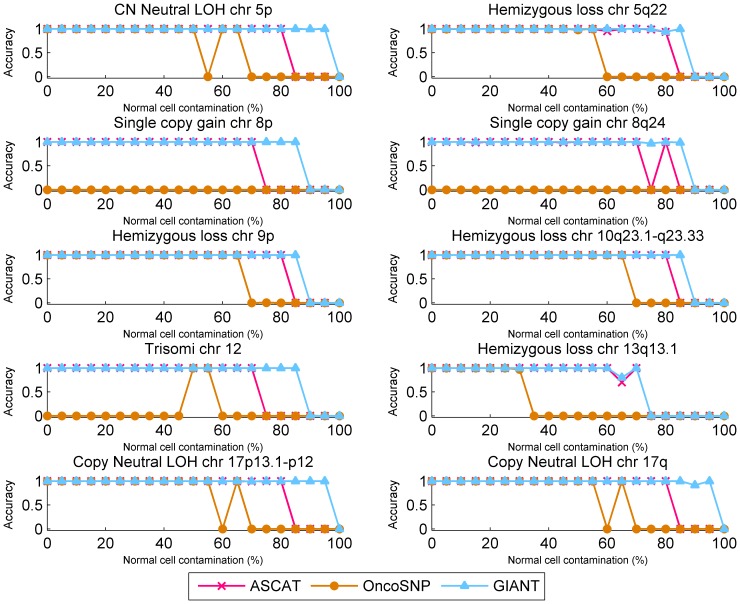
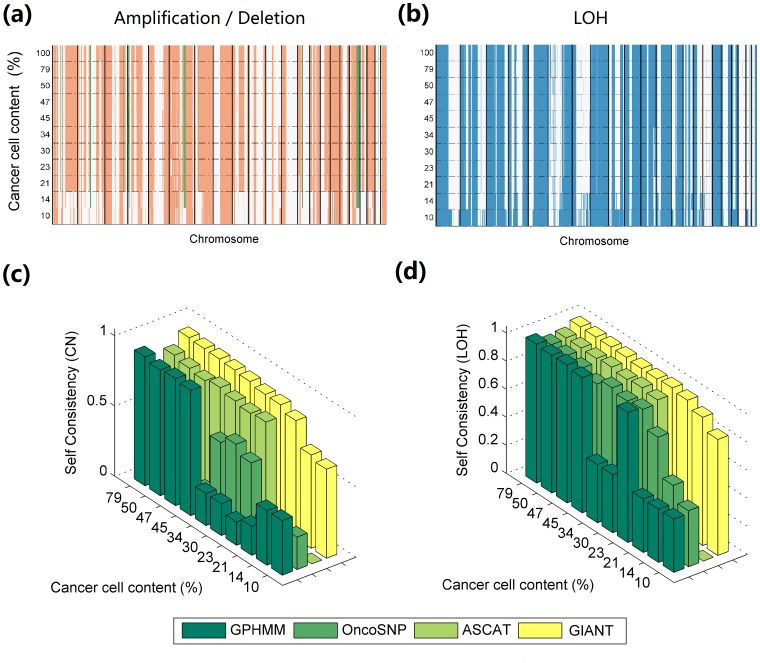
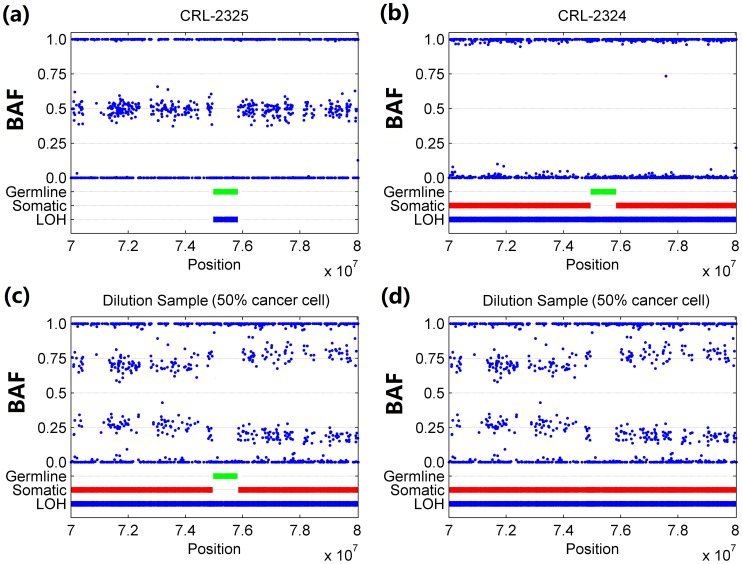
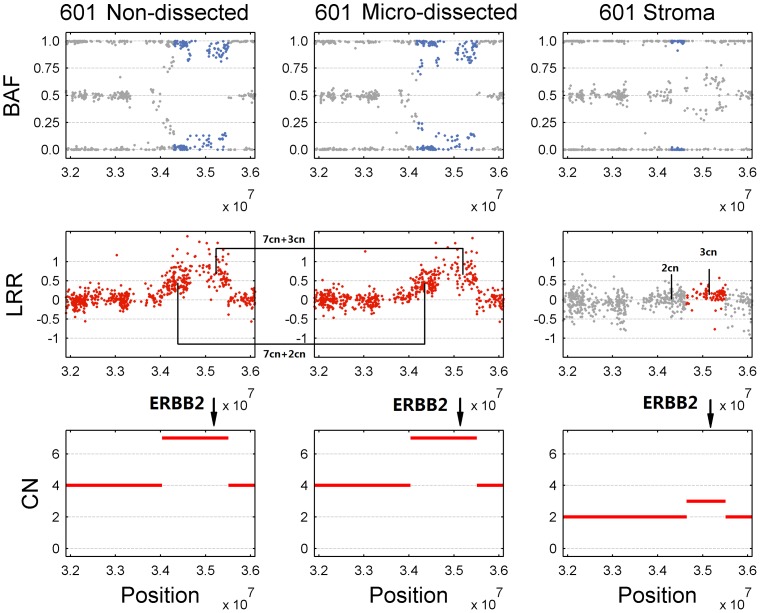
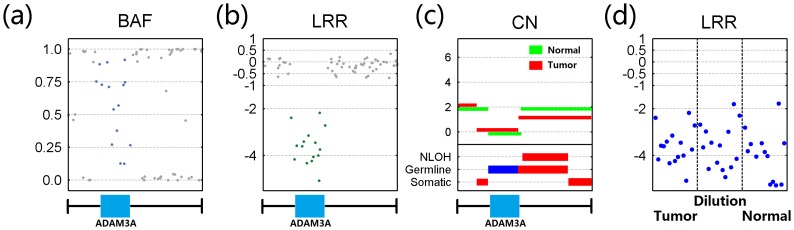
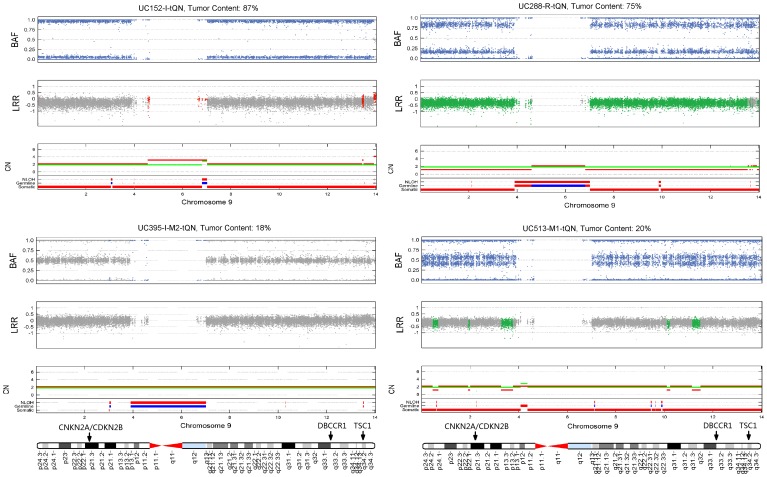
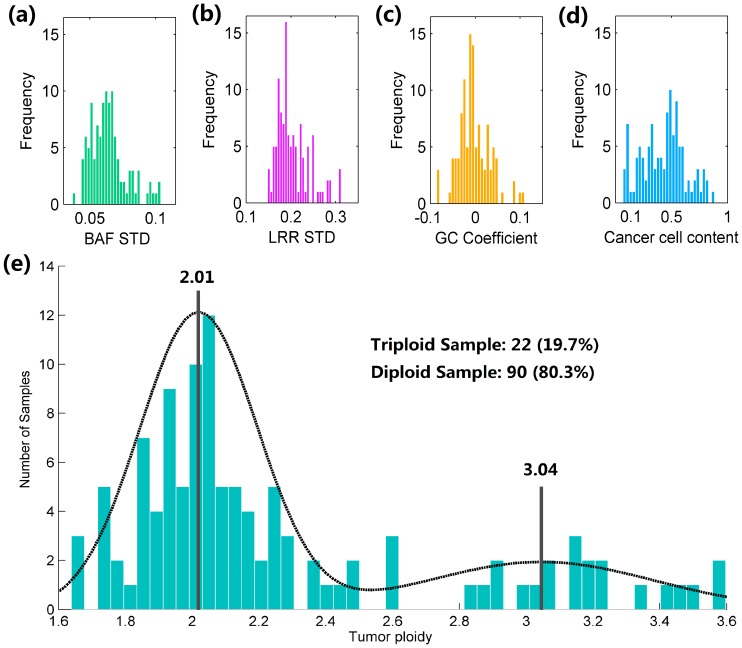
Genomic copy number alteration and allelic imbalance are distinct features of cancer cells, and recent advances in the genotyping technology have greatly boosted the research in the cancer genome. However, the complicated nature of tumor usually hampers the dissection of the SNP arrays. In this study, we describe a bioinformatic tool, named GIANT, for genome-wide identification of somatic aberrations from paired normal-tumor samples measured with SNP arrays. By efficiently incorporating genotype information of matched normal sample, it accurately detects different types of aberrations in cancer genome, even for aneuploid tumor samples with severe normal cell contamination. Furthermore, it allows for discovery of recurrent aberrations with critical biological properties in tumorigenesis by using statistical significance test. We demonstrate the superior performance of the proposed method on various datasets including tumor replicate pairs, simulated SNP arrays and dilution series of normal-cancer cell lines. Results show that GIANT has the potential to detect the genomic aberration even when the cancer cell proportion is as low as 5∼10%. Application on a large number of paired tumor samples delivers a genome-wide profile of the statistical significance of the various aberrations, including amplification, deletion and LOH. We believe that GIANT represents a powerful bioinformatic tool for interpreting the complex genomic aberration, and thus assisting both academic study and the clinical treatment of cancer.
基因组拷贝数改变和等位基因失衡是癌细胞的显著特征,并且基因分型技术的最新进展极大地推动了癌症基因组研究。然而,肿瘤的复杂性通常阻碍了对SNP阵列的剖析。在本研究中,我们描述了一种名为GIANT的生物信息学工具,用于从用SNP阵列测量的配对正常-肿瘤样本中进行全基因组范围的体细胞畸变鉴定。通过有效地整合匹配正常样本的基因型信息,它能准确检测癌症基因组中的不同类型畸变,即使对于具有严重正常细胞污染的非整倍体肿瘤样本也是如此。此外,它通过使用统计显著性检验,能够发现肿瘤发生过程中具有关键生物学特性的复发性畸变。我们在包括肿瘤重复对、模拟SNP阵列和正常-癌细胞系稀释系列在内的各种数据集上展示了所提出方法的卓越性能。结果表明,即使癌细胞比例低至5%至10%,GIANT也有检测基因组畸变的潜力。在大量配对肿瘤样本上的应用提供了全基因组范围的各种畸变(包括扩增、缺失和杂合性丢失)统计显著性概况。我们相信,GIANT是一种强大的生物信息学工具,可用于解释复杂的基因组畸变,从而辅助癌症的学术研究和临床治疗。