Yu Zhenhua, Li Ao, Wang Minghui
School of Information Science and Technology, University of Science and Technology of China, Hefei, AH230027, China.
Centers for Biomedical Engineering, University of Science and Technology of China, Hefei, AH230027, China.
BMC Med Genomics. 2017 Mar 15;10(1):15. doi: 10.1186/s12920-017-0255-4.
Copy number alterations (CNA) and loss of heterozygosity (LOH) represent a large proportion of genetic structural variations of cancer genomes. These aberrations are continuously accumulated during the procedure of clonal evolution and patterned by phylogenetic branching. This invariably results in the emergence of multiple cell populations with distinct complement of mutational landscapes in tumor sample. With the advent of next-generation sequencing technology, inference of subclonal populations has become one of the focused interests in cancer-associated studies, and is usually based on the assessment of combinations of somatic single-nucleotide variations (SNV), CNA and LOH. However, cancer samples often have several inherent issues, such as contamination of normal stroma, tumor aneuploidy and intra-tumor heterogeneity. Addressing these critical issues is imperative for accurate profiling of clonal architecture.
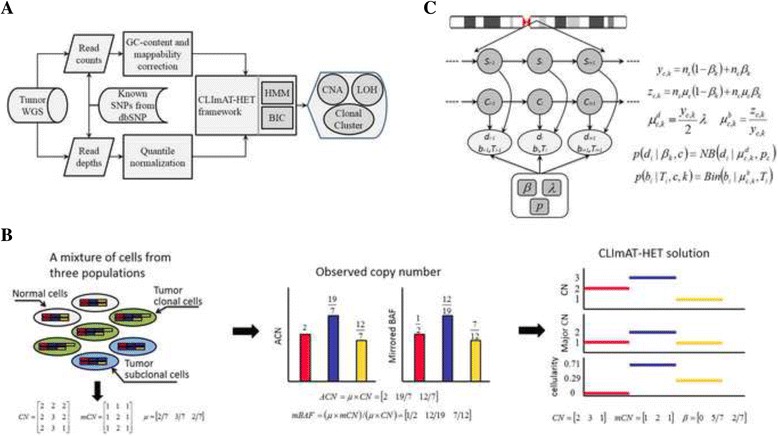
We present CLImAT-HET, a computational method designed for capturing clonal diversity in the CNA/LOH dimensions by taking into account the intra-tumor heterogeneity issue, in the case where a reference or matched normal sample is absent. The algorithm quantitatively represents the clonal identification problem using a factorial hidden Markov model, and takes an integrated analysis of read counts and allele frequency data. It is able to infer subclonal CNA and LOH events as well as the fraction of cells harboring each event.
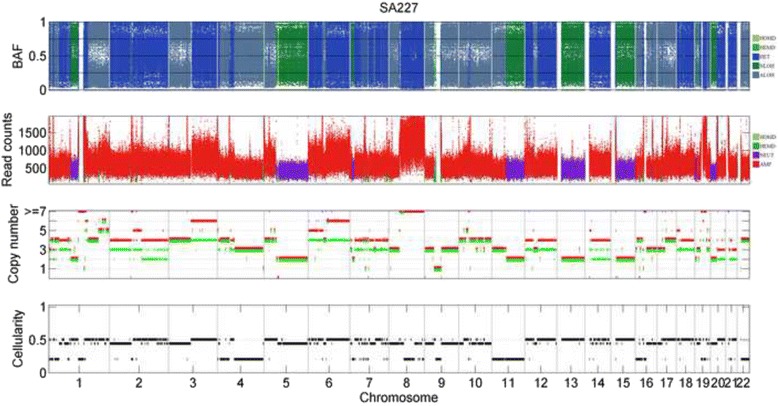
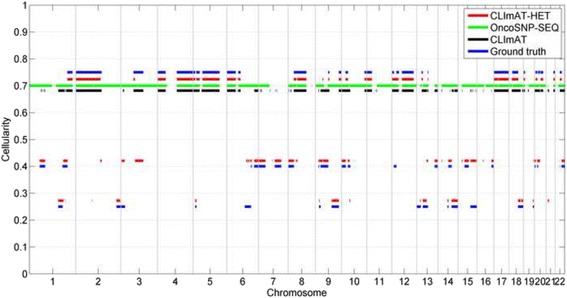
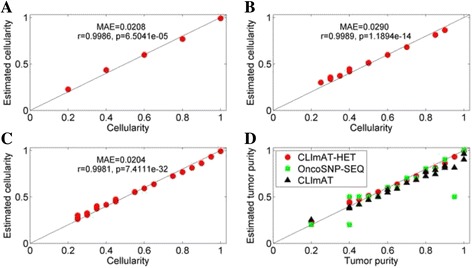
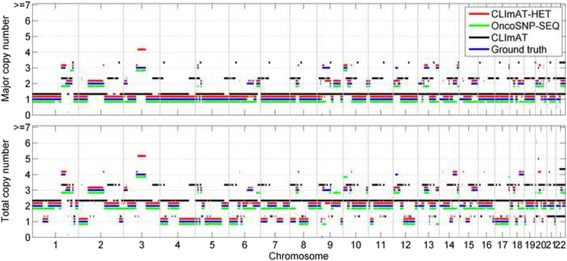
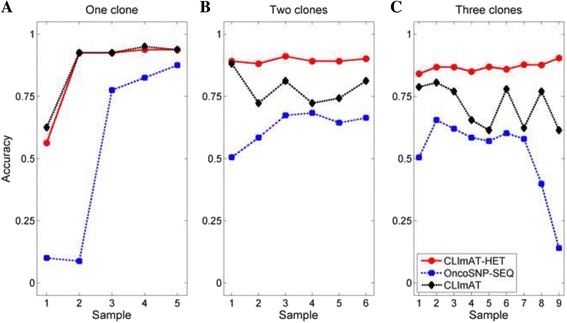
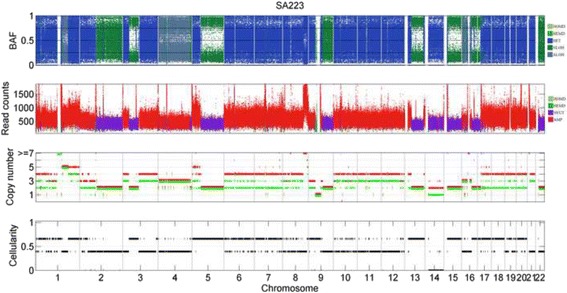
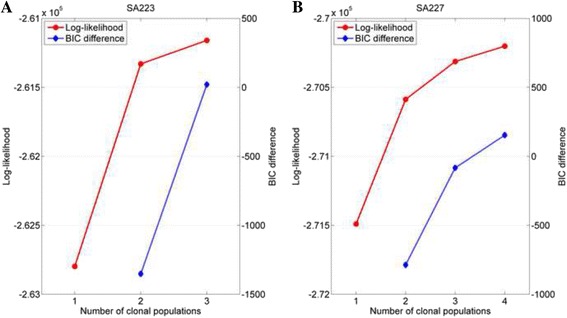
The results on simulated datasets indicate that CLImAT-HET has high power to identify CNA/LOH segments, it achieves an average accuracy of 0.87. It can also accurately infer proportion of each clonal population with an overall Pearson correlation coefficient of 0.99 and a mean absolute error of 0.02. CLImAT-HET shows significant advantages when compared with other existing methods. Application of CLImAT-HET to 5 primary triple negative breast cancer samples demonstrates its ability to capture clonal diversity in the CAN/LOH dimensions. It detects two clonal populations in one sample, and three clonal populations in one other sample.
CLImAT-HET, a novel algorithm is introduced to infer CNA/LOH segments from heterogeneous tumor samples. We demonstrate CLImAT-HET's ability to accurately recover clonal compositions using tumor WGS data without a match normal sample.
拷贝数改变(CNA)和杂合性缺失(LOH)占癌症基因组遗传结构变异的很大比例。这些畸变在克隆进化过程中不断积累,并由系统发育分支形成模式。这必然导致肿瘤样本中出现具有不同突变景观互补的多个细胞群体。随着下一代测序技术的出现,亚克隆群体的推断已成为癌症相关研究的重点关注之一,通常基于对体细胞单核苷酸变异(SNV)、CNA和LOH组合的评估。然而,癌症样本往往存在几个固有问题,如正常基质的污染、肿瘤非整倍性和肿瘤内异质性。解决这些关键问题对于准确描绘克隆结构至关重要。
我们提出了CLImAT-HET,这是一种计算方法,旨在通过考虑肿瘤内异质性问题,在没有参考或匹配正常样本的情况下,在CNA/LOH维度中捕获克隆多样性。该算法使用因子隐马尔可夫模型定量表示克隆识别问题,并对读取计数和等位基因频率数据进行综合分析。它能够推断亚克隆CNA和LOH事件以及携带每个事件的细胞比例。
模拟数据集的结果表明,CLImAT-HET具有很高的识别CNA/LOH片段的能力,平均准确率达到0.87。它还可以准确推断每个克隆群体的比例,总体皮尔逊相关系数为0.99,平均绝对误差为0.02。与其他现有方法相比,CLImAT-HET显示出显著优势。将CLImAT-HET应用于5个原发性三阴性乳腺癌样本,证明了其在CAN/LOH维度中捕获克隆多样性的能力。它在一个样本中检测到两个克隆群体,在另一个样本中检测到三个克隆群体。
引入了一种新算法CLImAT-HET,用于从异质性肿瘤样本中推断CNA/LOH片段。我们证明了CLImAT-HET能够使用肿瘤全基因组测序(WGS)数据准确恢复克隆组成,而无需匹配的正常样本。