Miga Karen H, Newton Yulia, Jain Miten, Altemose Nicolas, Willard Huntington F, Kent W James
Duke Institute for Genome Sciences & Policy, Duke University, Durham, North Carolina 27708, USA;
Genome Res. 2014 Apr;24(4):697-707. doi: 10.1101/gr.159624.113. Epub 2014 Feb 5.
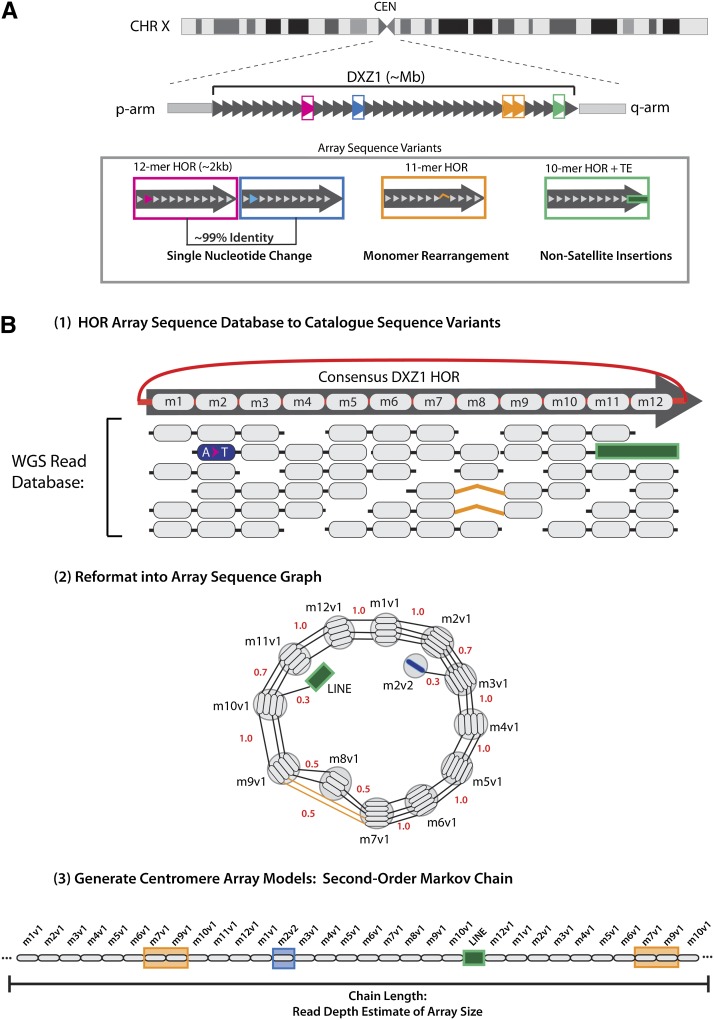
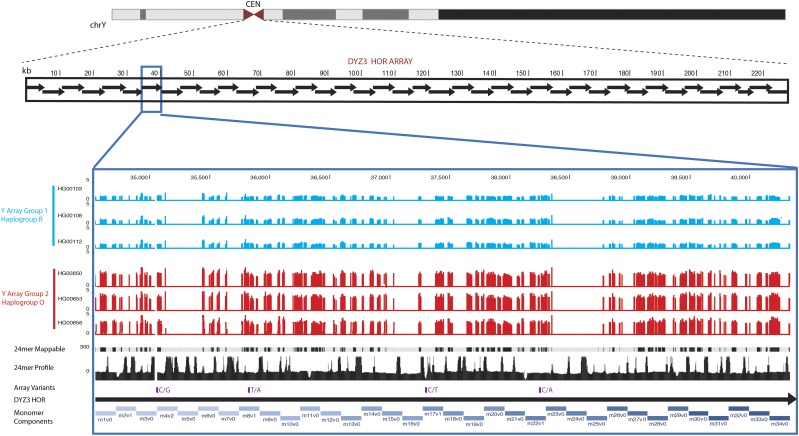
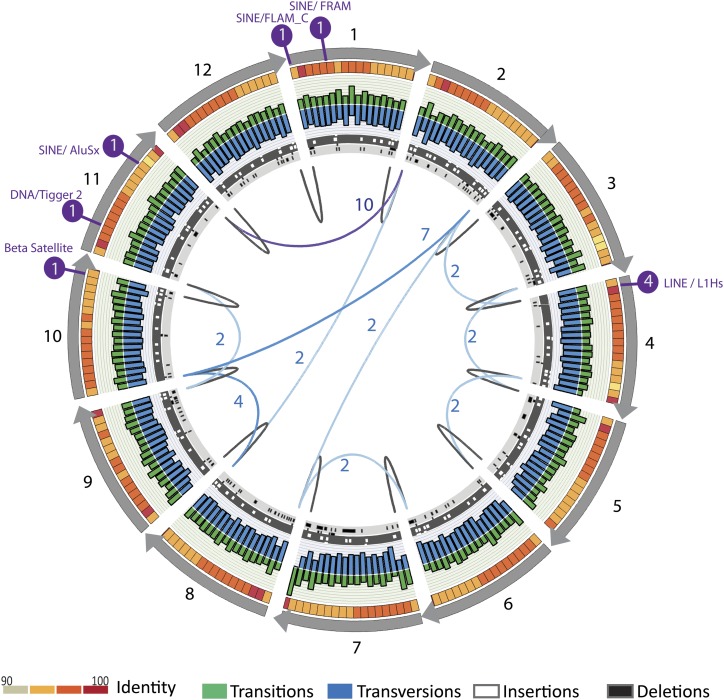
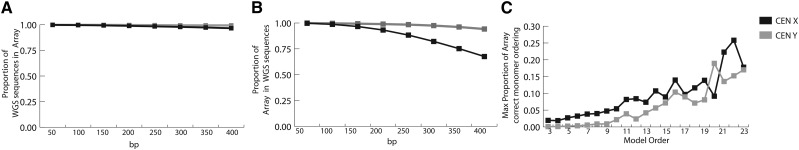
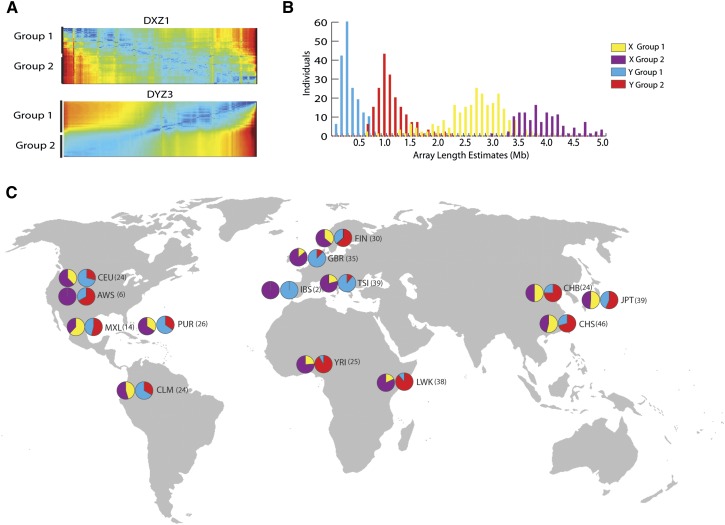
The human genome sequence remains incomplete, with multimegabase-sized gaps representing the endogenous centromeres and other heterochromatic regions. Available sequence-based studies within these sites in the genome have demonstrated a role in centromere function and chromosome pairing, necessary to ensure proper chromosome segregation during cell division. A common genomic feature of these regions is the enrichment of long arrays of near-identical tandem repeats, known as satellite DNAs, which offer a limited number of variant sites to differentiate individual repeat copies across millions of bases. This substantial sequence homogeneity challenges available assembly strategies and, as a result, centromeric regions are omitted from ongoing genomic studies. To address this problem, we utilize monomer sequence and ordering information obtained from whole-genome shotgun reads to model two haploid human satellite arrays on chromosomes X and Y, resulting in an initial characterization of 3.83 Mb of centromeric DNA within an individual genome. To further expand the utility of each centromeric reference sequence model, we evaluate sites within the arrays for short-read mappability and chromosome specificity. Because satellite DNAs evolve in a concerted manner, we use these centromeric assemblies to assess the extent of sequence variation among 366 individuals from distinct human populations. We thus identify two satellite array variants in both X and Y centromeres, as determined by array length and sequence composition. This study provides an initial sequence characterization of a regional centromere and establishes a foundation to extend genomic characterization to these sites as well as to other repeat-rich regions within complex genomes.
人类基因组序列仍不完整,存在代表内源性着丝粒和其他异染色质区域的多兆碱基大小的缺口。基因组中这些位点内现有的基于序列的研究表明,它们在着丝粒功能和染色体配对中发挥作用,这对于确保细胞分裂期间染色体的正确分离是必要的。这些区域的一个常见基因组特征是富含长串几乎相同的串联重复序列,即卫星DNA,它们提供的变异位点数量有限,难以区分数百万碱基上的各个重复拷贝。这种高度的序列同质性给现有的组装策略带来了挑战,因此着丝粒区域被排除在正在进行的基因组研究之外。为了解决这个问题,我们利用从全基因组鸟枪法测序读数中获得的单体序列和排序信息,对X和Y染色体上的两个单倍体人类卫星阵列进行建模,从而对个体基因组内383万个碱基的着丝粒DNA进行了初步表征。为了进一步扩展每个着丝粒参考序列模型的实用性,我们评估了阵列内的位点的短读长可映射性和染色体特异性。由于卫星DNA以协同方式进化,我们使用这些着丝粒组装体来评估来自不同人类群体的366个个体之间的序列变异程度。因此,我们确定了X和Y着丝粒中的两个卫星阵列变体,这是由阵列长度和序列组成决定的。这项研究提供了区域着丝粒的初步序列特征,并为将基因组特征扩展到这些位点以及复杂基因组中其他富含重复序列的区域奠定了基础。