Panwar Bharat, Arora Amit, Raghava Gajendra P S
Bioinformatics Centre, Institute of Microbial Technology (CSIR), Sector 39A, Chandigarh, India.
BMC Genomics. 2014 Feb 13;15:127. doi: 10.1186/1471-2164-15-127.
Evidence is accumulating that non-coding transcripts, previously thought to be functionally inert, play important roles in various cellular activities. High throughput techniques like next generation sequencing have resulted in the generation of vast amounts of sequence data. It is therefore desirable, not only to discriminate coding and non-coding transcripts, but also to assign the noncoding RNA (ncRNA) transcripts into respective classes (families). Although there are several algorithms available for this task, their classification performance remains a major concern. Acknowledging the crucial role that non-coding transcripts play in cellular processes, it is required to develop algorithms that are able to precisely classify ncRNA transcripts.
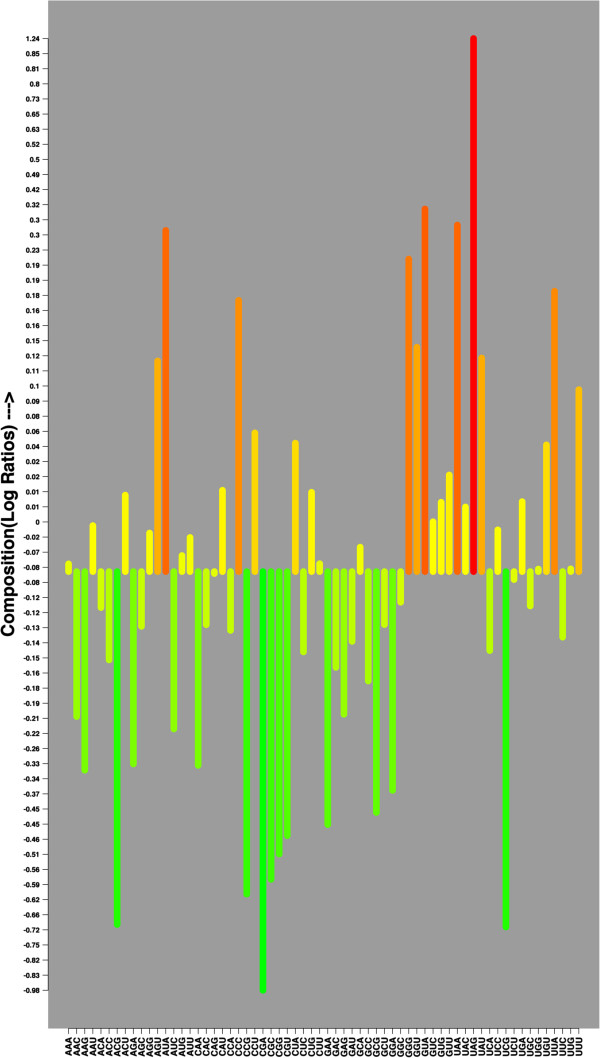
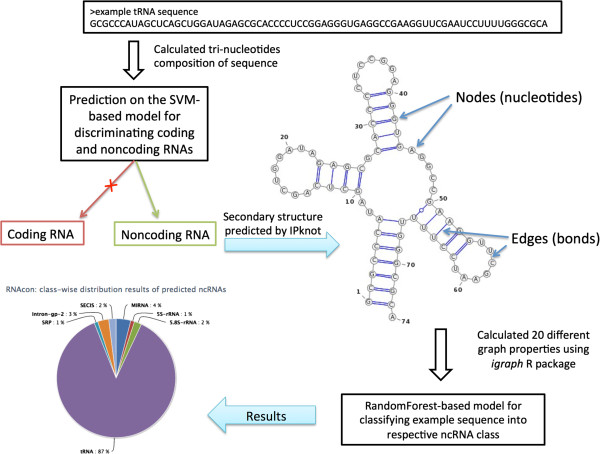
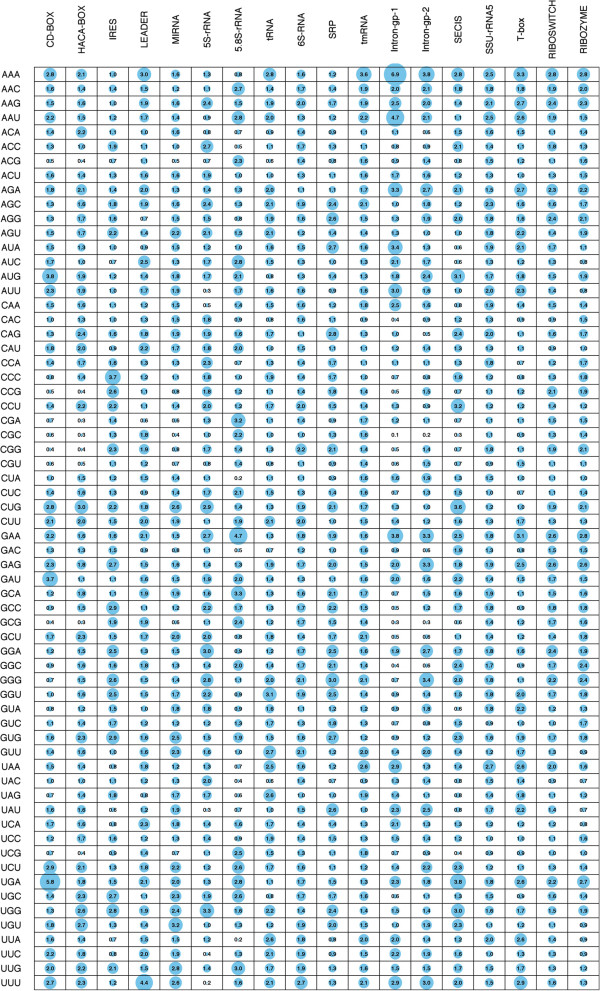
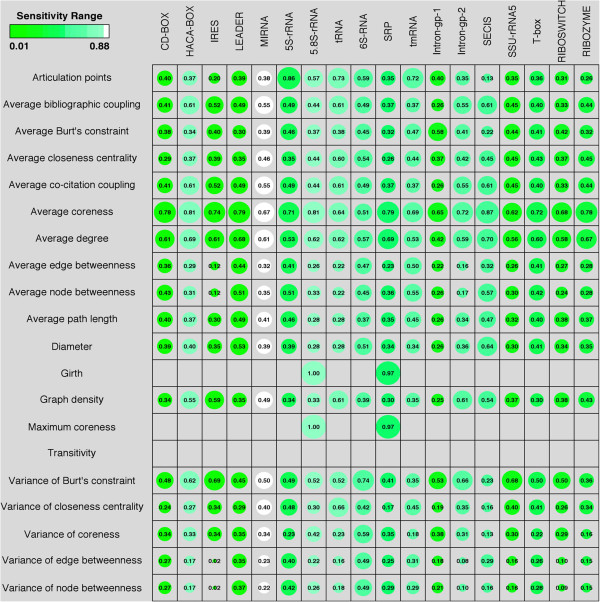
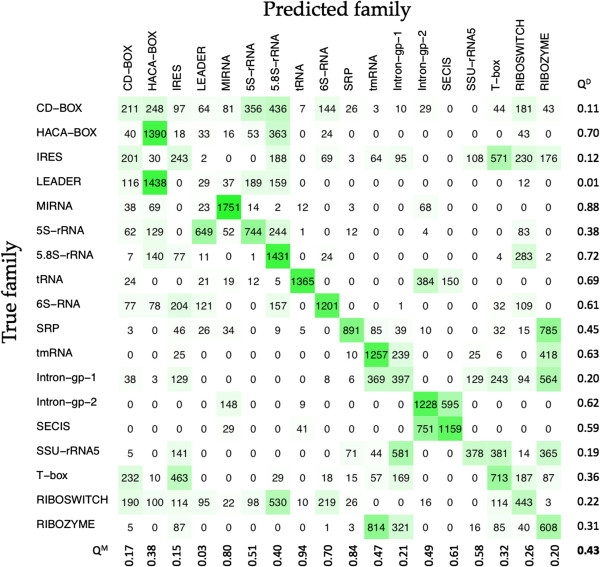
In this study, we initially develop prediction tools to discriminate coding or non-coding transcripts and thereafter classify ncRNAs into respective classes. In comparison to the existing methods that employed multiple features, our SVM-based method by using a single feature (tri-nucleotide composition), achieved MCC of 0.98. Knowing that the structure of a ncRNA transcript could provide insights into its biological function, we use graph properties of predicted ncRNA structures to classify the transcripts into 18 different non-coding RNA classes. We developed classification models using a variety of algorithms (BayeNet, NaiveBayes, MultilayerPerceptron, IBk, libSVM, SMO and RandomForest) and observed that model based on RandomForest performed better than other models. As compared to the GraPPLE study, the sensitivity (of 13 classes) and specificity (of 14 classes) was higher. Moreover, the overall sensitivity of 0.43 outperforms the sensitivity of GraPPLE (0.33) whereas the overall MCC measure of 0.40 (in contrast to MCC of 0.29 of GraPPLE) was significantly higher for our method. This clearly demonstrates that our models are more accurate than existing models.
This work conclusively demonstrates that a simple feature, tri-nucleotide composition, is sufficient to discriminate between coding and non-coding RNA sequences. Similarly, graph properties based feature set along with RandomForest algorithm are most suitable to classify different ncRNA classes. We have also developed an online and standalone tool-- RNAcon ( http://crdd.osdd.net/raghava/rnacon).
越来越多的证据表明,以前被认为功能惰性的非编码转录本在各种细胞活动中发挥着重要作用。像下一代测序这样的高通量技术已经产生了大量的序列数据。因此,不仅需要区分编码和非编码转录本,还需要将非编码RNA(ncRNA)转录本归类到各自的类别(家族)中。虽然有几种算法可用于此任务,但其分类性能仍然是一个主要问题。认识到非编码转录本在细胞过程中所起的关键作用,需要开发能够精确分类ncRNA转录本的算法。
在本研究中,我们首先开发了预测工具来区分编码或非编码转录本,然后将ncRNA分类到各自的类别中。与采用多种特征的现有方法相比,我们基于支持向量机(SVM)的方法通过使用单个特征(三核苷酸组成),马修斯相关系数(MCC)达到了0.98。由于知道ncRNA转录本的结构可以为其生物学功能提供见解,我们使用预测的ncRNA结构的图形属性将转录本分类为18种不同的非编码RNA类别。我们使用多种算法(贝叶斯网络、朴素贝叶斯、多层感知器、IBk、libSVM、SMO和随机森林)开发了分类模型,并观察到基于随机森林的模型比其他模型表现更好。与GraPPLE研究相比,(13个类别的)灵敏度和(14个类别的)特异性更高。此外,0.43的总体灵敏度优于GraPPLE的灵敏度(0.33),而我们方法的总体MCC值为0.40(相比之下,GraPPLE的MCC为0.29)则显著更高。这清楚地表明我们的模型比现有模型更准确。
这项工作最终证明了一个简单的特征,即三核苷酸组成,足以区分编码和非编码RNA序列。同样,基于图形属性的特征集与随机森林算法最适合对不同的ncRNA类别进行分类。我们还开发了一个在线和独立的工具——RNAcon(http://crdd.osdd.net/raghava/rnacon)。