Lady Davis Institute for Medical Research, Jewish General Hospital Montreal, QC, Canada ; Department of Epidemiology, Biostatistics and Occupational Health, McGill University Montreal, QC, Canada.
Department of Epidemiology, Biostatistics and Occupational Health, McGill University Montreal, QC, Canada.
Front Genet. 2014 Jan 29;5:11. doi: 10.3389/fgene.2014.00011. eCollection 2014.
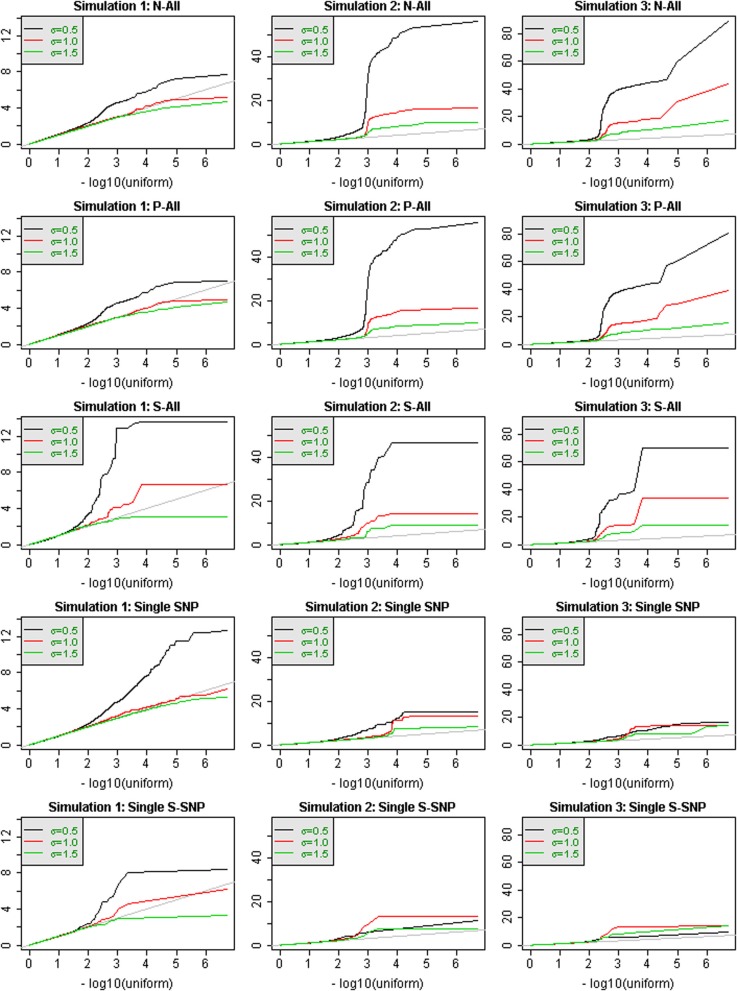
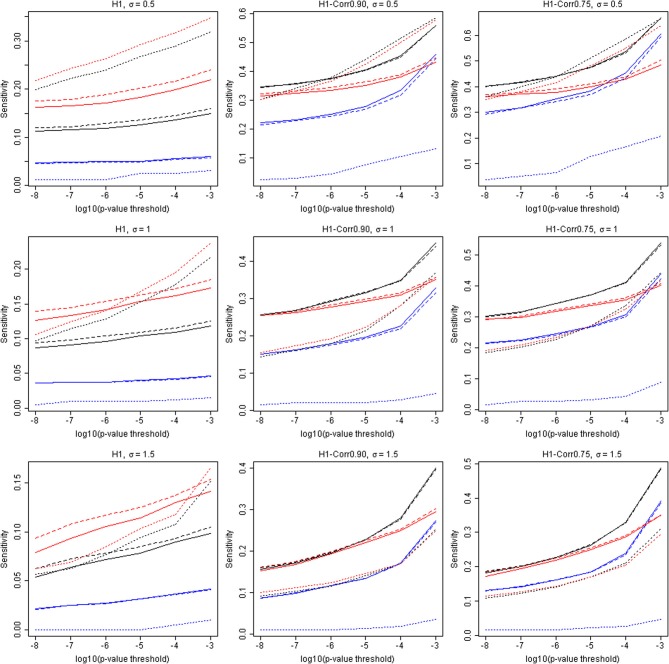
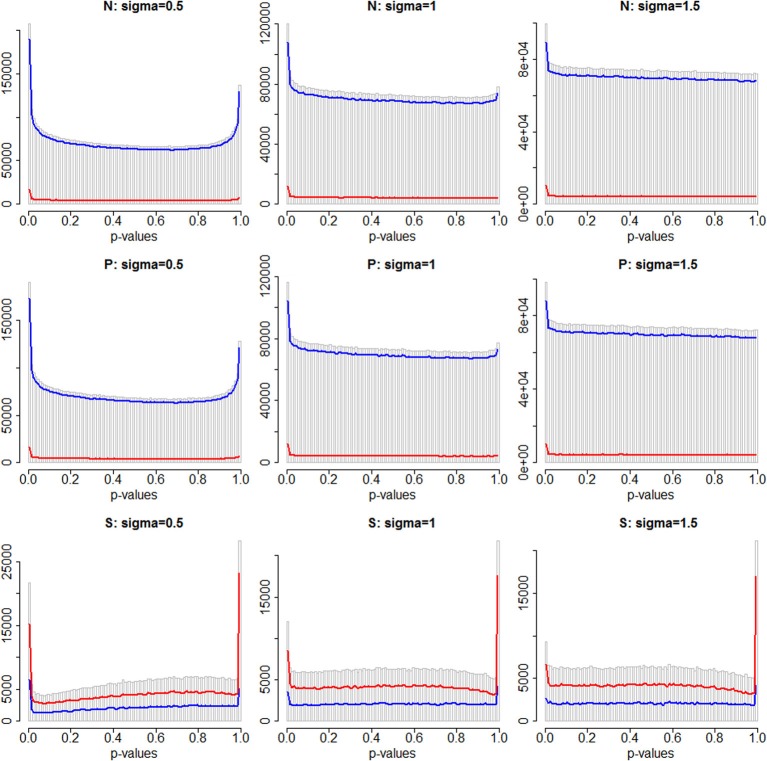
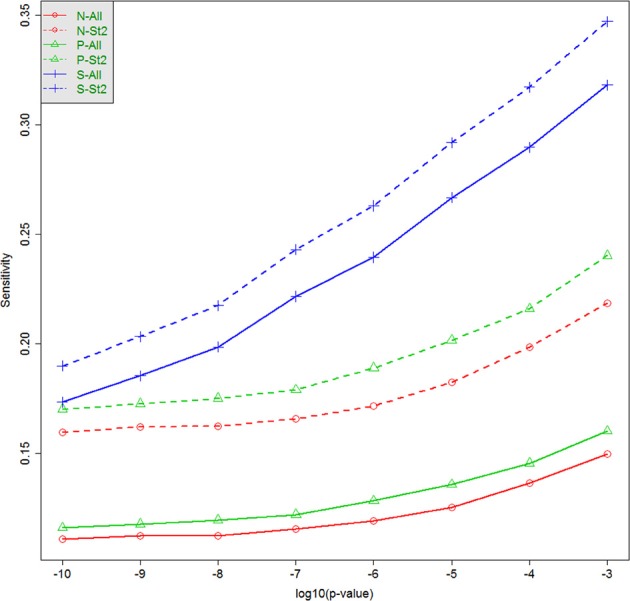
When analyzing the data that arises from exome or whole-genome sequencing studies, window-based tests, (i.e., tests that jointly analyze all genetic data in a small genomic region), are very popular. However, power is known to be quite low for finding associations with phenotypes using these tests, and therefore a variety of analytic strategies may be employed to potentially improve power. Using sequencing data of all of chromosome 3 from an interim release of data on 2432 individuals from the UK10K project, we simulated phenotypes associated with rare genetic variation, and used the results to explore the window-based test power. We asked two specific questions: firstly, whether there could be substantial benefits associated with incorporating information from external annotation on the genetic variants, and secondly whether the false discovery rate (FDRs) would be a useful metric for assessing significance. Although, as expected, there are benefits to using additional information (such as annotation) when it is associated with causality, we confirmed the general pattern of low sensitivity and power for window-based tests. For our chosen example, even when power is high to detect some of the associations, many of the regions containing causal variants are not detectable, despite using lax significance thresholds and optimal analytic methods. Furthermore, our estimated FDR values tended to be much smaller than the true FDRs. Long-range correlations between variants-due to linkage disequilibrium-likely explain some of this bias. A more sophisticated approach to using the annotation information may improve power, however, many causal variants of realistic effect sizes may simply be undetectable, at least with this sample size. Perhaps annotation information could assist in distinguishing windows containing causal variants from windows that are merely correlated with causal variants.
在分析外显子组或全基因组测序研究产生的数据时,基于窗口的测试(即联合分析小基因组区域内所有遗传数据的测试)非常流行。然而,使用这些测试发现与表型相关联的关联的功效已知是相当低的,因此可以采用各种分析策略来提高潜在的功效。使用来自 UK10K 项目的 2432 个人的中间数据释放的所有染色体 3 的测序数据,我们模拟了与罕见遗传变异相关的表型,并使用结果来探索基于窗口的测试功效。我们提出了两个具体问题:首先,是否可以从遗传变异的外部注释中获得大量相关信息;其次,错误发现率(FDR)是否可以作为评估显着性的有用指标。尽管,如预期的那样,当附加信息(如注释)与因果关系相关时,会有好处,但我们确认了基于窗口的测试的敏感性和功效普遍较低的模式。对于我们选择的示例,即使在检测到一些关联的功效很高的情况下,许多包含因果变异的区域仍然无法检测到,尽管使用了宽松的显着性阈值和最佳分析方法。此外,我们估计的 FDR 值往往远小于真实 FDR 值。由于连锁不平衡导致的变异之间的长程相关性可能解释了部分偏差。使用注释信息的更复杂方法可能会提高功效,但是,许多现实效应大小的因果变异可能根本无法检测到,至少在这种样本量下是如此。也许注释信息可以帮助区分包含因果变异的窗口和仅与因果变异相关的窗口。