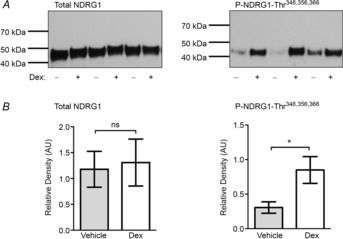
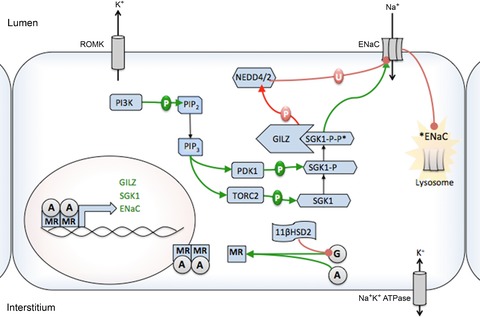
Hunter Robert W, Ivy Jessica R, Bailey Matthew A
University/BHF Centre for Cardiovascular Science, The Queen's Medical Research Institute, 47 Little France Crescent, Edinburgh EH16 4TJ, UK.
J Physiol. 2014 Apr 15;592(8):1731-44. doi: 10.1113/jphysiol.2013.267609. Epub 2014 Feb 17.
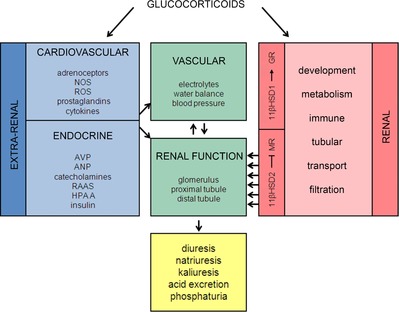
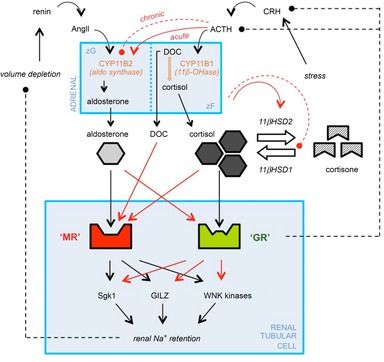
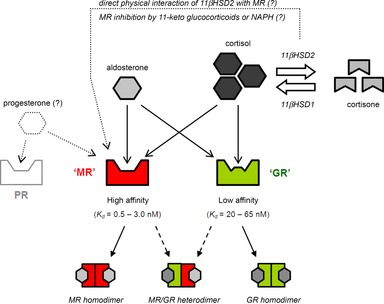
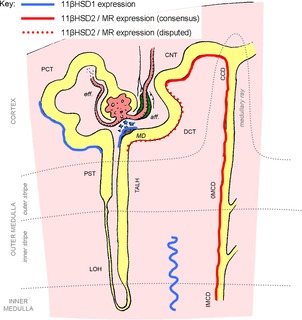
The clinical manifestations of glucocorticoid excess include central obesity, hyperglycaemia, dyslipidaemia, electrolyte abnormalities and hypertension. A century on from Cushing's original case study, these cardinal features are prevalent in industrialized nations. Hypertension is the major modifiable risk factor for cardiovascular and renal disease and reflects underlying abnormalities of Na(+) homeostasis. Aldosterone is a master regulator of renal Na(+) transport but here we argue that glucocorticoids are also influential, particularly during moderate excess. The hypothalamic-pituitary-adrenal axis can affect renal Na(+) homeostasis on multiple levels, systemically by increasing mineralocorticoid synthesis and locally by actions on both the mineralocorticoid and glucocorticoid receptors, both of which are expressed in the kidney. The kidney also expresses both of the 11β-hydroxysteroid dehydrogenase (11βHSD) enzymes. The intrarenal generation of active glucocorticoid by 11βHSD1 stimulates Na(+) reabsorption; failure to downregulate the enzyme during adaption to high dietary salt causes salt-sensitive hypertension. The deactivation of glucocorticoid by 11βHSD2 underpins the regulatory dominance for Na(+) transport of mineralocorticoids and defines the 'aldosterone-sensitive distal nephron'. In summary, glucocorticoids can stimulate renal transport processes conventionally attributed to the renin-angiotensin-aldosterone system. Importantly, Na(+) and volume homeostasis do not exert negative feedback on the hypothalamic-pituitary-adrenal axis. These actions are therefore clinically relevant and may contribute to the pathogenesis of hypertension in conditions associated with elevated glucocorticoid levels, such as the metabolic syndrome and chronic stress.
糖皮质激素过多的临床表现包括向心性肥胖、高血糖、血脂异常、电解质紊乱和高血压。自库欣最初的病例研究过去一个世纪以来,这些主要特征在工业化国家普遍存在。高血压是心血管和肾脏疾病的主要可改变风险因素,反映了钠(Na⁺)稳态的潜在异常。醛固酮是肾脏钠(Na⁺)转运的主要调节因子,但我们认为糖皮质激素也有影响,尤其是在中度过量时。下丘脑 - 垂体 - 肾上腺轴可在多个层面影响肾脏钠(Na⁺)稳态,在全身通过增加盐皮质激素合成,在局部通过作用于盐皮质激素和糖皮质激素受体,这两种受体均在肾脏中表达。肾脏还表达两种11β - 羟基类固醇脱氢酶(11βHSD)。11βHSD1在肾脏内产生活性糖皮质激素可刺激钠(Na⁺)重吸收;在适应高盐饮食期间未能下调该酶会导致盐敏感性高血压。11βHSD2使糖皮质激素失活,这是盐皮质激素对钠(Na⁺)转运具有调节优势的基础,并定义了“醛固酮敏感远端肾单位”。总之,糖皮质激素可刺激传统上归因于肾素 - 血管紧张素 - 醛固酮系统的肾脏转运过程。重要的是,钠(Na⁺)和容量稳态不会对下丘脑 - 垂体 - 肾上腺轴产生负反馈。因此,这些作用在临床上具有相关性,可能有助于与糖皮质激素水平升高相关的疾病(如代谢综合征和慢性应激)中高血压的发病机制。