Singh Sharon A, Goldberg Tracie A, Henson Adrianna L, Husain-Krautter Sehba, Nihrane Abdallah, Blanc Lionel, Ellis Steven R, Lipton Jeffrey M, Liu Johnson M
Department of Molecular Medicine, Hofstra North Shore-LIJ School of Medicine, Hempstead, New York, United States of America ; The Feinstein Institute for Medical Research, Manhasset, New York, United States of America ; Division of Hematology/Oncology, Steven and Alexandra Cohen Children's Medical Center of New York, New Hyde Park, New York, United States of America ; Department of Pediatrics, Hofstra North Shore-LIJ School of Medicine, Hempstead, New York, United States of America.
The Feinstein Institute for Medical Research, Manhasset, New York, United States of America ; Division of Hematology/Oncology, Steven and Alexandra Cohen Children's Medical Center of New York, New Hyde Park, New York, United States of America.
PLoS One. 2014 Feb 18;9(2):e89098. doi: 10.1371/journal.pone.0089098. eCollection 2014.
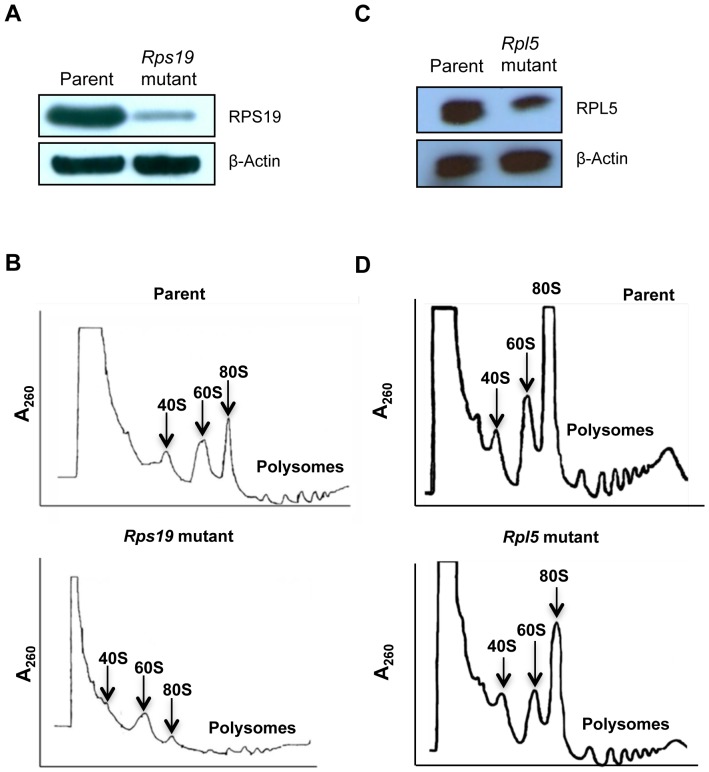
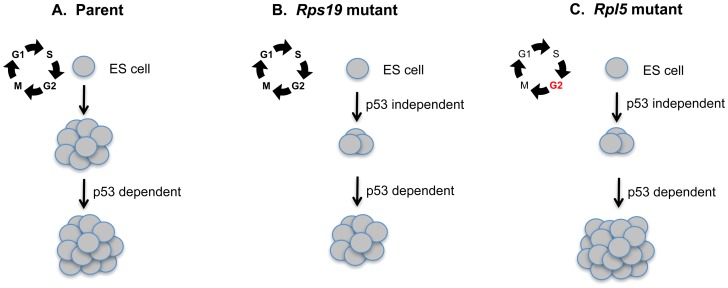
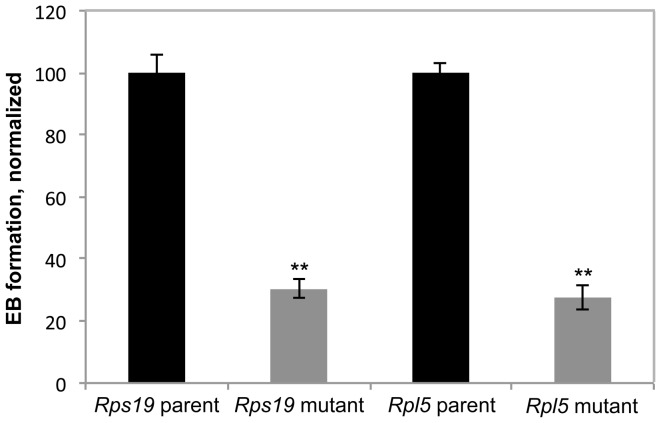
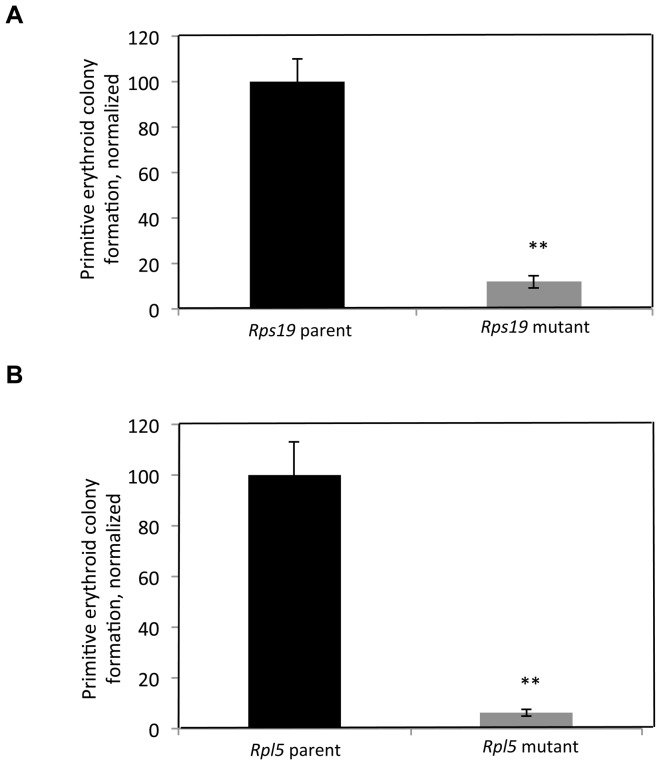
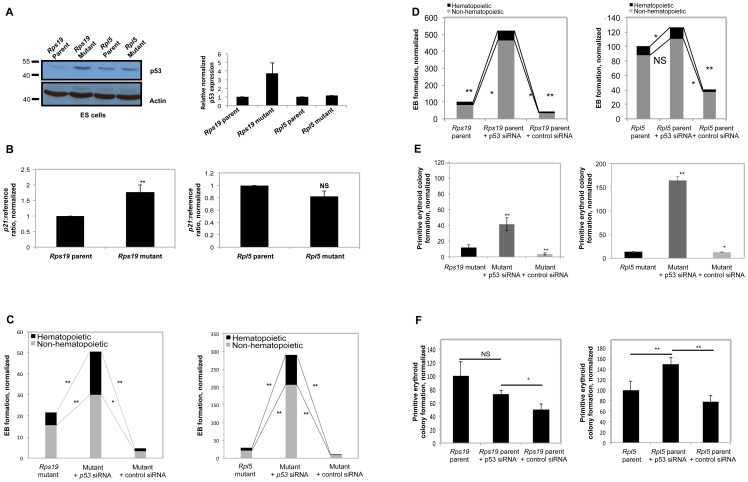
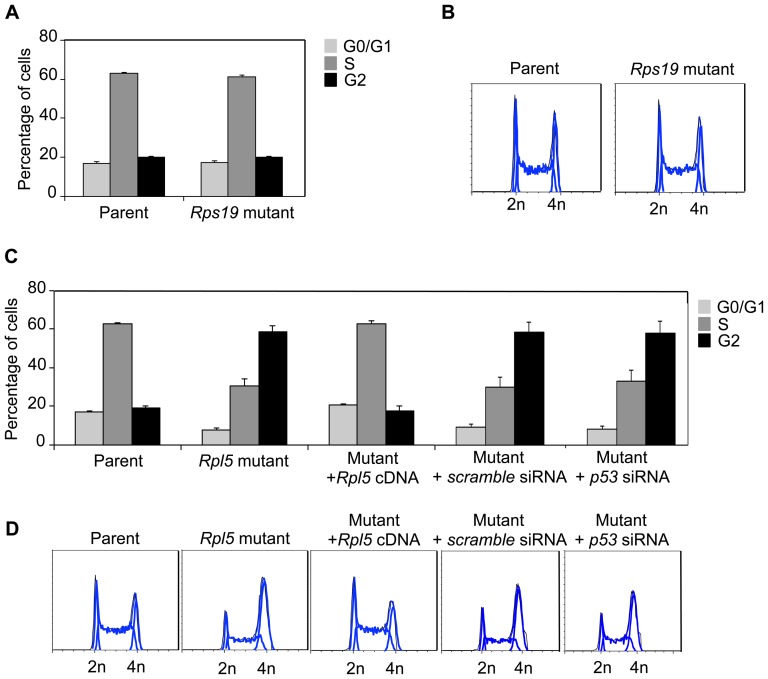
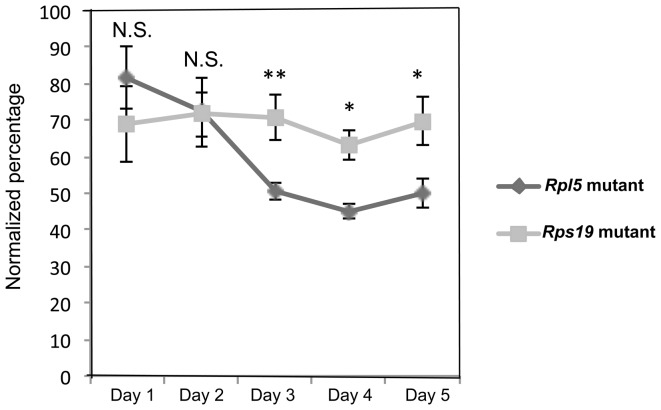
Diamond Blackfan anemia (DBA) is a rare inherited bone marrow failure syndrome caused by ribosomal protein haploinsufficiency. DBA exhibits marked phenotypic variability, commonly presenting with erythroid hypoplasia, less consistently with non-erythroid features. The p53 pathway, activated by abortive ribosome assembly, is hypothesized to contribute to the erythroid failure of DBA. We studied murine embryonic stem (ES) cell lines harboring a gene trap mutation in a ribosomal protein gene, either Rps19 or Rpl5. Both mutants exhibited ribosomal protein haploinsufficiency and polysome defects. Rps19 mutant ES cells showed significant increase in p53 protein expression, however, there was no similar increase in the Rpl5 mutant cells. Embryoid body formation was diminished in both mutants but nonspecifically rescued by knockdown of p53. When embryoid bodies were further differentiated to primitive erythroid colonies, both mutants exhibited a marked reduction in colony formation, which was again nonspecifically rescued by p53 inhibition. Cell cycle analyses were normal in Rps19 mutant ES cells, but there was a significant delay in the G2/M phase in the Rpl5 mutant cells, which was unaffected by p53 knockdown. Concordantly, Rpl5 mutant ES cells had a more pronounced growth defect in liquid culture compared to the Rps19 mutant cells. We conclude that the defects in our RPS19 and RPL5 haploinsufficient mouse ES cells are not adequately explained by p53 stabilization, as p53 knockdown appears to increase the growth and differentiation potential of both parental and mutant cells. Our studies demonstrate that gene trap mouse ES cells are useful tools to study the pathogenesis of DBA.
先天性纯红细胞再生障碍性贫血(DBA)是一种由核糖体蛋白单倍体不足引起的罕见遗传性骨髓衰竭综合征。DBA表现出明显的表型变异性,通常表现为红系发育不全,较少出现非红系特征。由流产性核糖体组装激活的p53通路被认为与DBA的红系衰竭有关。我们研究了在核糖体蛋白基因Rps19或Rpl5中存在基因陷阱突变的小鼠胚胎干细胞(ES)系。两种突变体均表现出核糖体蛋白单倍体不足和多核糖体缺陷。Rps19突变的ES细胞中p53蛋白表达显著增加,然而,Rpl5突变细胞中没有类似的增加。两种突变体的胚状体形成均减少,但通过敲低p53可非特异性地挽救。当胚状体进一步分化为原始红系集落时,两种突变体的集落形成均显著减少,这同样可通过抑制p53非特异性地挽救。Rps19突变的ES细胞的细胞周期分析正常,但Rpl5突变细胞在G2/M期有明显延迟,这不受p53敲低的影响。一致地,与Rps19突变细胞相比,Rpl5突变的ES细胞在液体培养中有更明显的生长缺陷。我们得出结论,p53稳定化不能充分解释我们的RPS19和RPL5单倍体不足小鼠ES细胞中的缺陷,因为敲低p53似乎增加了亲本细胞和突变细胞的生长及分化潜能。我们的研究表明,基因陷阱小鼠ES细胞是研究DBA发病机制的有用工具。