Gérard Francine C A, Yang Ruifeng, Romani Bizhan, Poisson Alexis, Belzile Jean-Philippe, Rougeau Nicole, Cohen Éric A
Institut de Recherches Cliniques de Montréal (IRCM), Montréal, Québec, Canada.
Institut de Recherches Cliniques de Montréal (IRCM), Montréal, Québec, Canada ; Department of Microbiology, Infectiology and Immunology, Université de Montréal, Montréal, Québec, Canada.
PLoS One. 2014 Feb 18;9(2):e89195. doi: 10.1371/journal.pone.0089195. eCollection 2014.
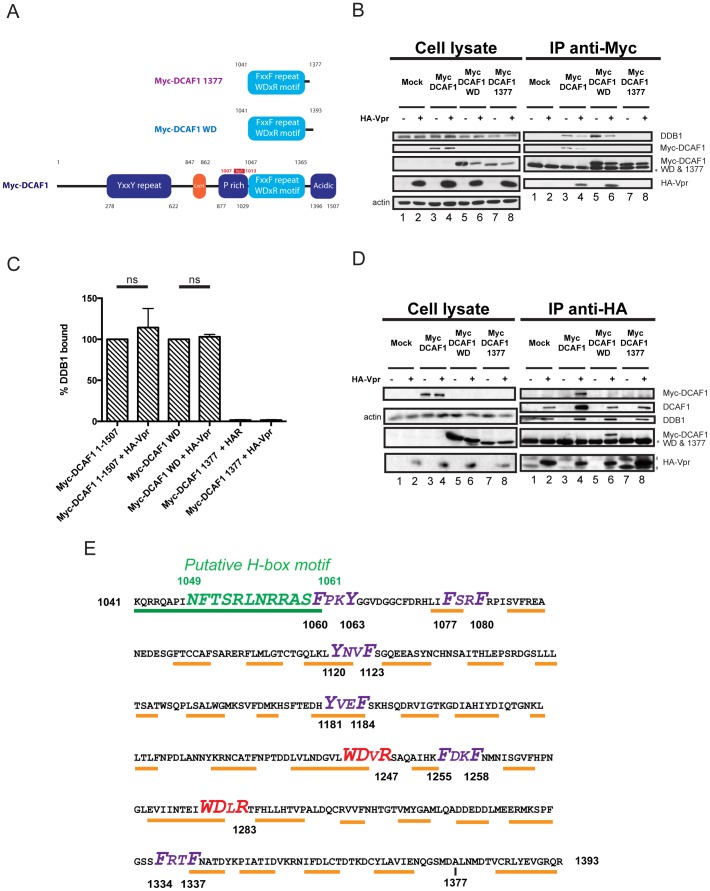
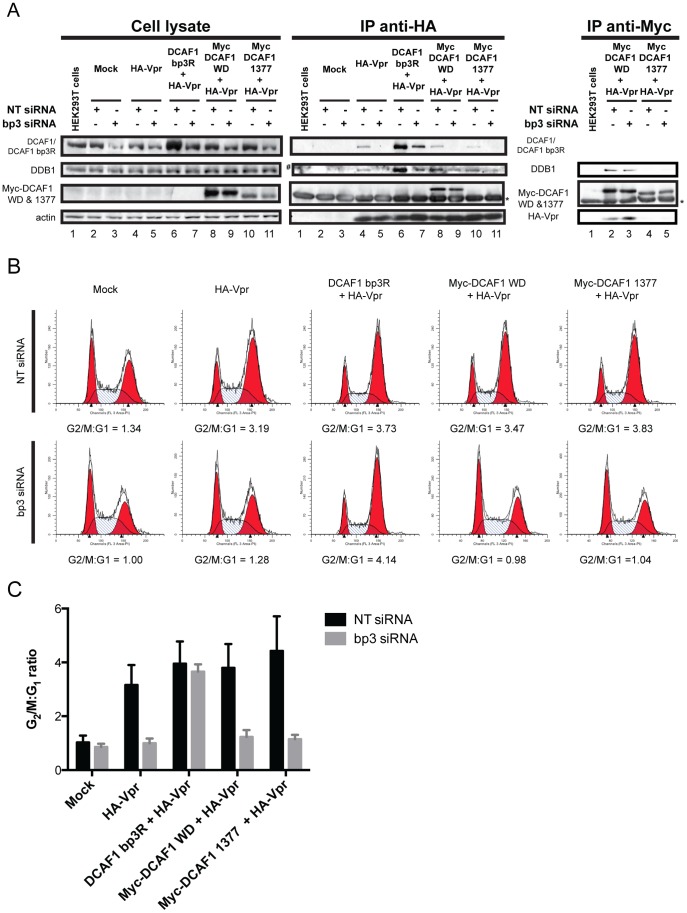
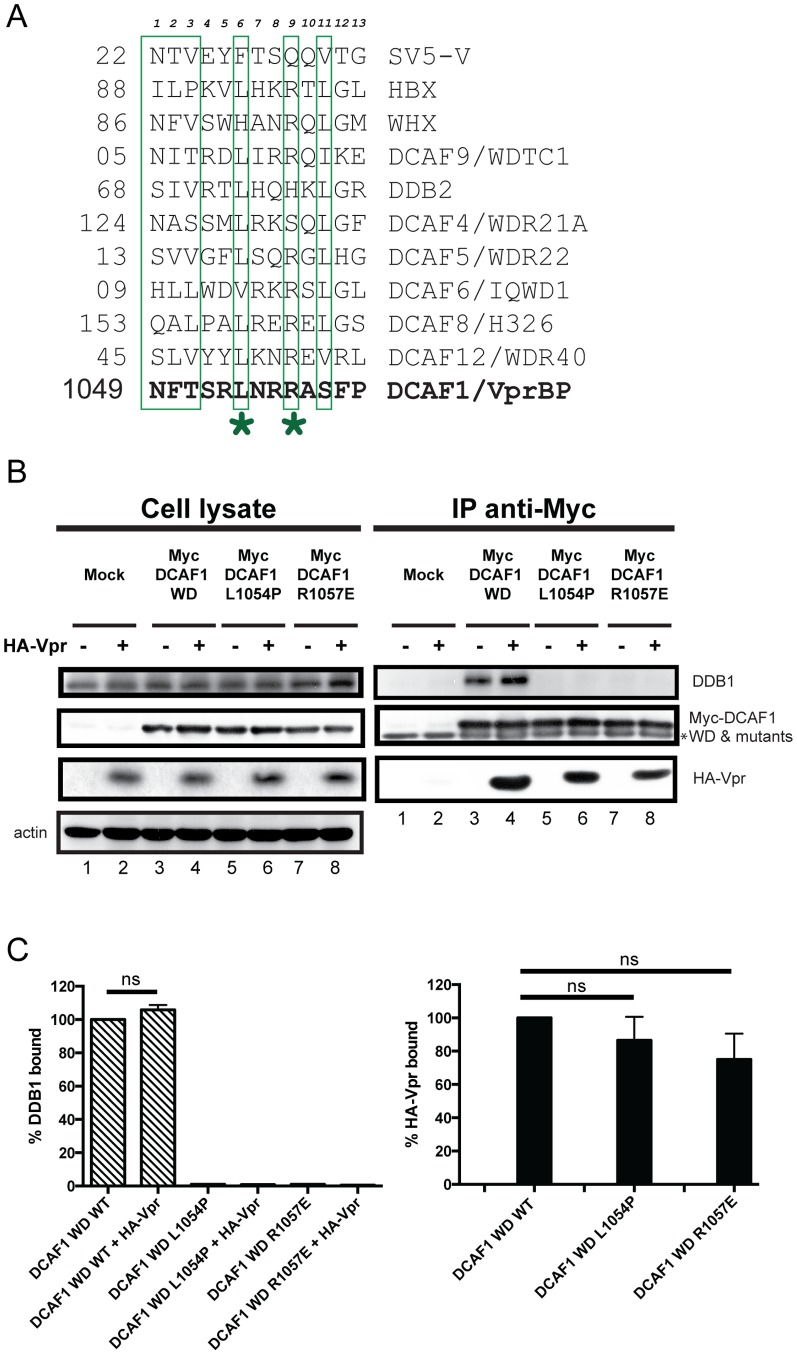
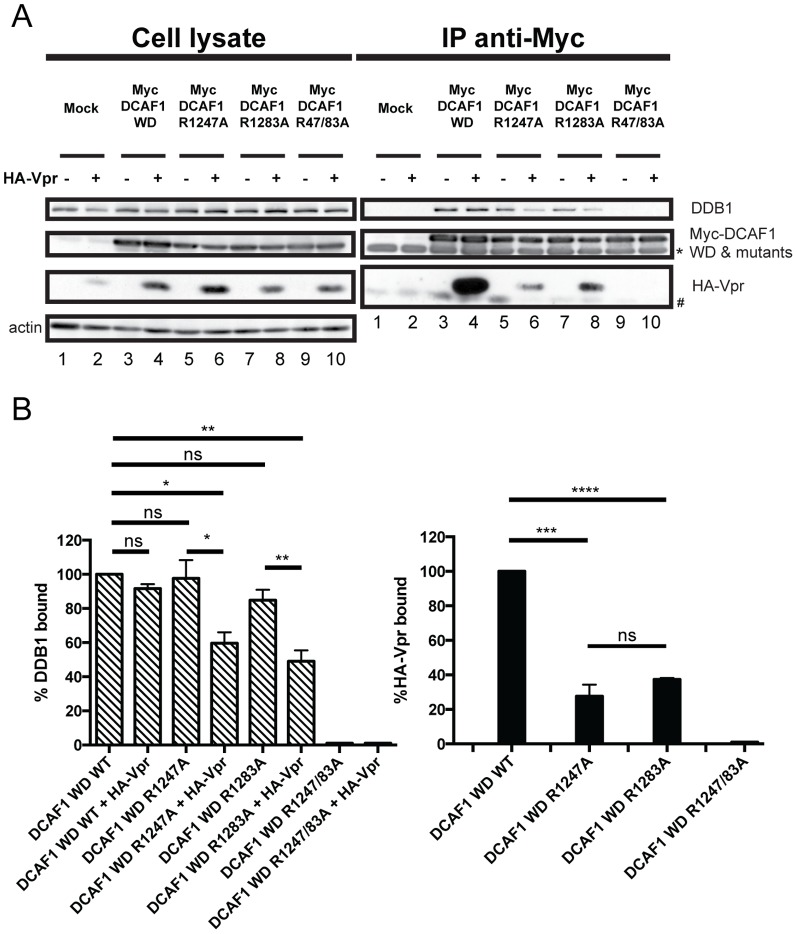
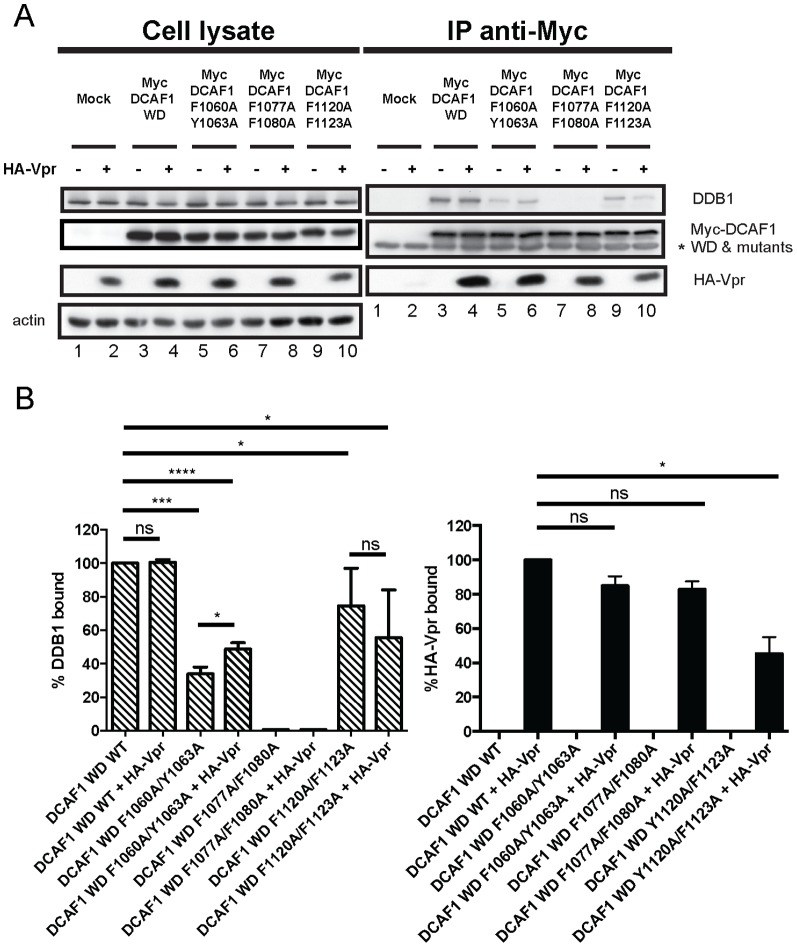
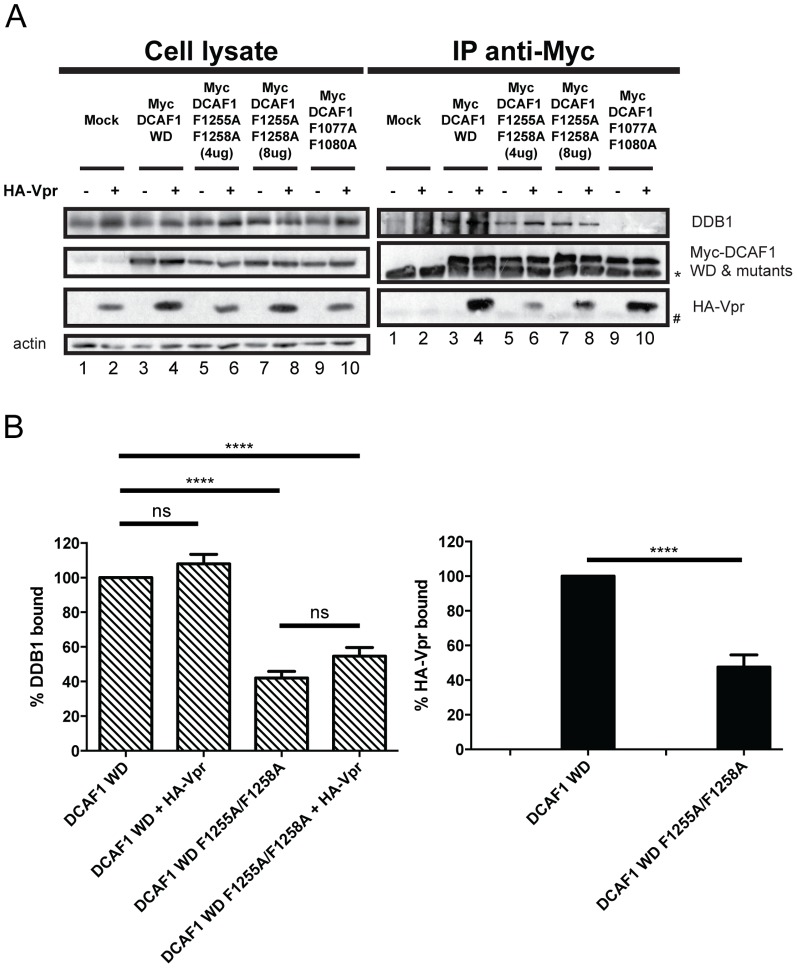
HIV viral protein R (Vpr) induces a cell cycle arrest at the G2/M phase by activating the ATR DNA damage/replication stress signalling pathway through engagement of the DDB1-CUL4A-DCAF1 E3 ubiquitin ligase via a direct binding to the substrate specificity receptor DCAF1. Since no high resolution structures of the DDB1-DCAF1-Vpr substrate recognition module currently exist, we used a mutagenesis approach to better define motifs in DCAF1 that are crucial for Vpr and DDB1 binding. Herein, we show that the minimal domain of DCAF1 that retained the ability to bind Vpr and DDB1 was mapped to residues 1041 to 1393 (DCAF1 WD). Mutagenic analyses identified an α-helical H-box motif and F/YxxF/Y motifs located in the N-terminal domain of DCAF1 WD that are involved in exclusive binding to DDB1. While we could not identify elements specifically involved in Vpr binding, overall, the mutagenesis data suggest that the predicted β-propeller conformation of DCAF1 is likely to be critical for Vpr association. Importantly, we provide evidence that binding of Vpr to DCAF1 appears to modulate the formation of a DDB1/DCAF1 complex. Lastly, we show that expression of DCAF1 WD in the absence of endogenous DCAF1 was not sufficient to enable Vpr-mediated G2 arrest activity. Overall, our results reveal that Vpr and DDB1 binding on DCAF1 can be genetically separated and further suggest that DCAF1 contains determinants in addition to the Vpr and DDB1 minimal binding domain, which are required for Vpr to enable the induction of a G2 arrest.
HIV病毒蛋白R(Vpr)通过与底物特异性受体DCAF1直接结合,参与DDB1-CUL4A-DCAF1 E3泛素连接酶的作用,激活ATR DNA损伤/复制应激信号通路,从而诱导细胞周期在G2/M期停滞。由于目前尚无DDB1-DCAF1-Vpr底物识别模块的高分辨率结构,我们采用诱变方法来更好地确定DCAF1中对Vpr和DDB1结合至关重要的基序。在此,我们表明,保留与Vpr和DDB1结合能力的DCAF1最小结构域定位于第1041至1393位残基(DCAF1 WD)。诱变分析确定了位于DCAF1 WD N端结构域的一个α螺旋H盒基序和F/YxxF/Y基序,它们参与与DDB1的特异性结合。虽然我们无法确定与Vpr结合的特异性元件,但总体而言,诱变数据表明DCAF1预测的β螺旋桨构象可能对Vpr结合至关重要。重要的是,我们提供证据表明Vpr与DCAF1的结合似乎调节了DDB1/DCAF1复合物的形成。最后,我们表明在缺乏内源性DCAF1的情况下,DCAF1 WD的表达不足以实现Vpr介导的G2期停滞活性。总体而言,我们的结果表明Vpr和DDB1在DCAF1上的结合可以在基因上分离,并且进一步表明DCAF1除了Vpr和DDB1最小结合结构域外还包含决定因素,这些因素是Vpr诱导G2期停滞所必需的。