Podsiadło Agnieszka, Wrzesień Mariusz, Paja Wiesław, Rudnicki Witold, Wilczyński Bartek
BMC Syst Biol. 2013;7 Suppl 6(Suppl 6):S16. doi: 10.1186/1752-0509-7-S6-S16. Epub 2013 Dec 13.
Transcriptional regulation in multi-cellular organisms is a complex process involving multiple modular regulatory elements for each gene. Building whole-genome models of transcriptional networks requires mapping all relevant enhancers and then linking them to target genes. Previous methods of enhancer identification based either on sequence information or on epigenetic marks have different limitations stemming from incompleteness of each of these datasets taken separately.
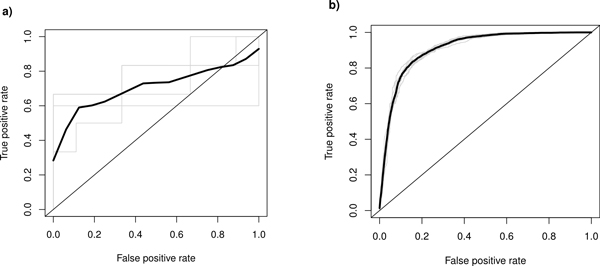
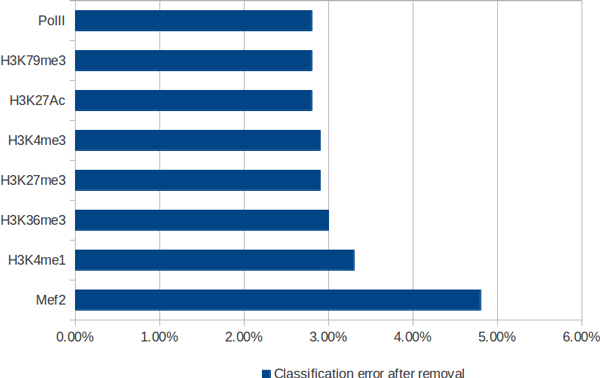
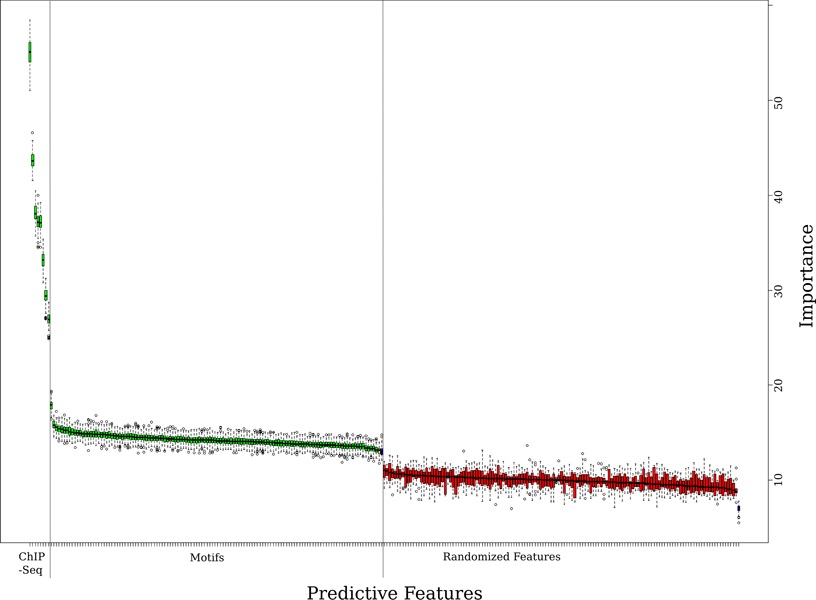
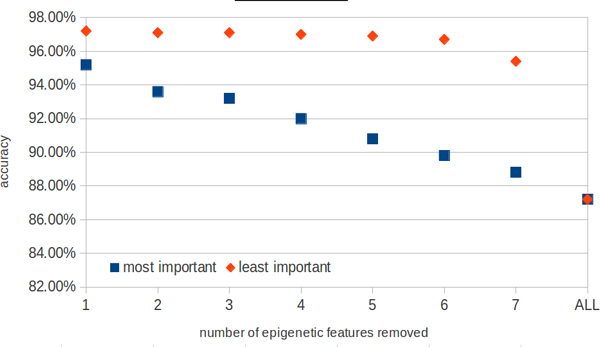
In this work we present a new approach for discovery of regulatory elements based on the combination of sequence motifs and epigenetic marks measured with ChIP-Seq. Our method uses supervised learning approaches to train a model describing the dependence of enhancer activity on sequence features and histone marks. Our results indicate that using combination of features provides superior results to previous approaches based on either one of the datasets. While histone modifications remain the dominant feature for accurate predictions, the models based on sequence motifs have advantages in their general applicability to different tissues. Additionally, we assess the relevance of different sequence motifs in prediction accuracy showing that even tissue-specific enhancer activity depends on multiple motifs.
Based on our results, we conclude that it is worthwhile to include sequence motif data into computational approaches to active enhancer prediction and also that classifiers trained on a specific set of enhancers can generalize with significant accuracy beyond the training set.
多细胞生物中的转录调控是一个复杂的过程,每个基因都涉及多个模块化调控元件。构建转录网络的全基因组模型需要绘制所有相关增强子,然后将它们与靶基因连接起来。以前基于序列信息或表观遗传标记的增强子识别方法,由于单独使用的每个数据集都不完整,存在不同的局限性。
在这项工作中,我们提出了一种基于序列基序和通过ChIP-Seq测量的表观遗传标记相结合的调控元件发现新方法。我们的方法使用监督学习方法来训练一个模型,该模型描述增强子活性对序列特征和组蛋白标记的依赖性。我们的结果表明,使用特征组合比基于单个数据集的先前方法提供了更好的结果。虽然组蛋白修饰仍然是准确预测的主要特征,但基于序列基序的模型在对不同组织的普遍适用性方面具有优势。此外,我们评估了不同序列基序在预测准确性中的相关性,表明即使是组织特异性增强子活性也依赖于多个基序。
基于我们的结果,我们得出结论,将序列基序数据纳入活跃增强子预测的计算方法是值得的,并且在一组特定增强子上训练的分类器可以以显著的准确性推广到训练集之外。