Li Wenyuan, Dai Chao, Kang Shuli, Zhou Xianghong Jasmine
Molecular and Computational Biology Program, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA.
Molecular and Computational Biology Program, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA.
Methods. 2014 Jun 1;67(3):313-24. doi: 10.1016/j.ymeth.2014.02.024. Epub 2014 Feb 28.
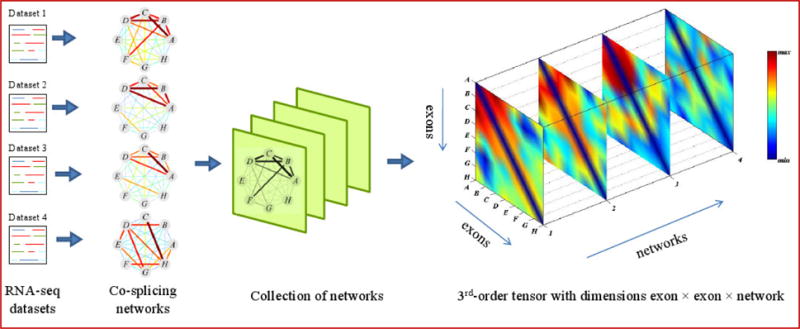
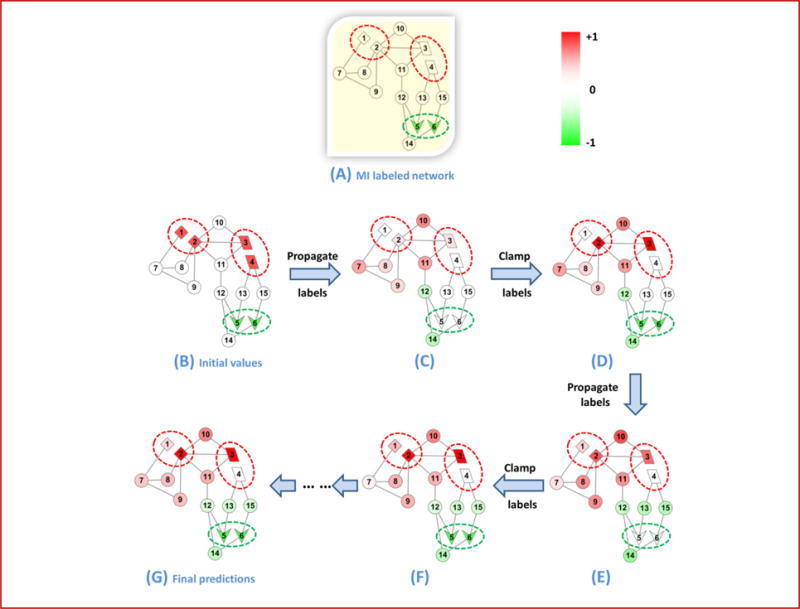
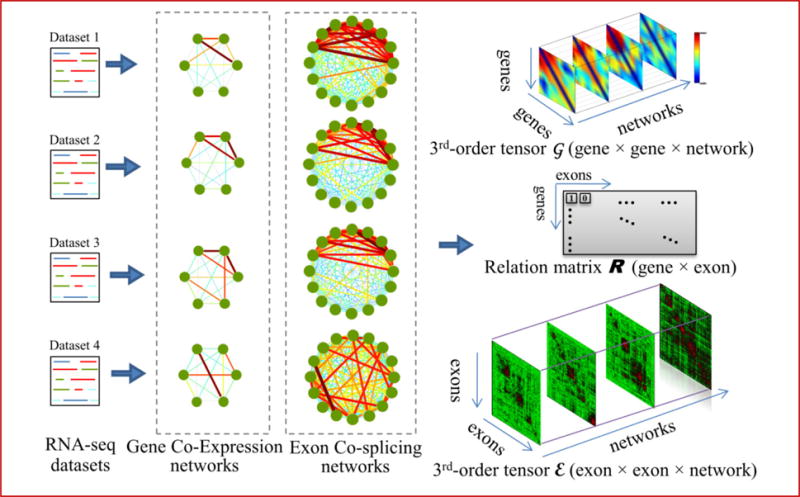
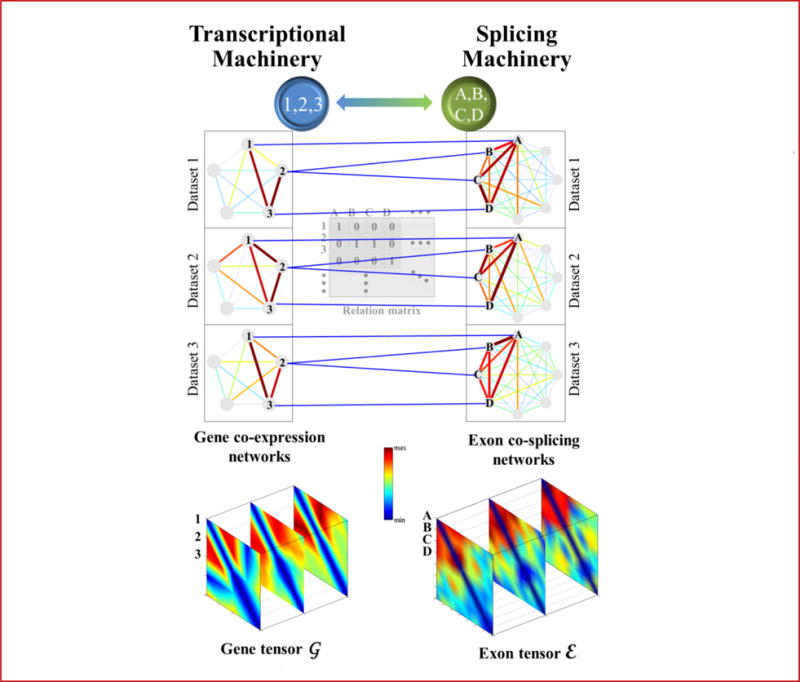
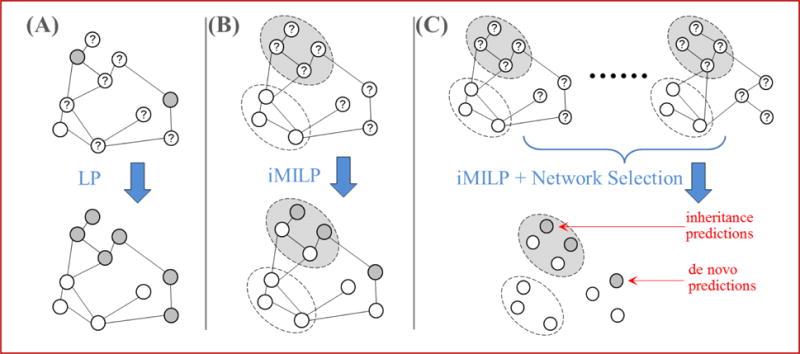
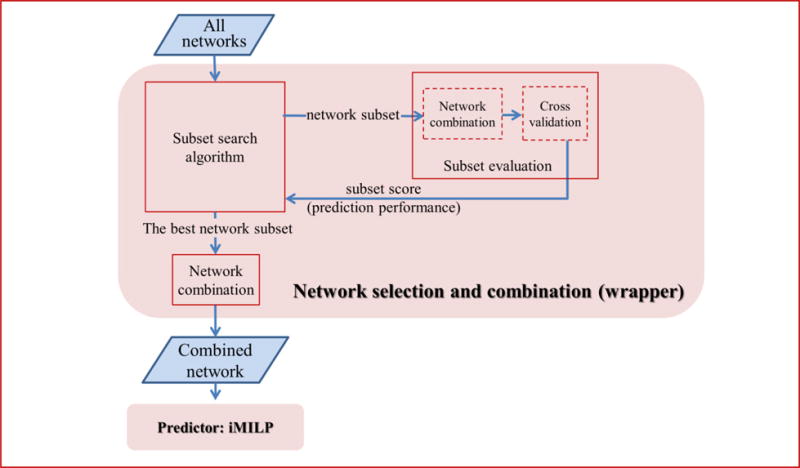
Alternative splicing is an important gene regulatory mechanism that dramatically increases the complexity of the proteome. However, how alternative splicing is regulated and how transcription and splicing are coordinated are still poorly understood, and functions of transcript isoforms have been studied only in a few limited cases. Nowadays, RNA-seq technology provides an exceptional opportunity to study alternative splicing on genome-wide scales and in an unbiased manner. With the rapid accumulation of data in public repositories, new challenges arise from the urgent need to effectively integrate many different RNA-seq datasets for study alterative splicing. This paper discusses a set of advanced computational methods that can integrate and analyze many RNA-seq datasets to systematically identify splicing modules, unravel the coupling of transcription and splicing, and predict the functions of splicing isoforms on a genome-wide scale.
可变剪接是一种重要的基因调控机制,它极大地增加了蛋白质组的复杂性。然而,可变剪接是如何被调控的,以及转录和剪接是如何协调的,目前仍知之甚少,并且转录本异构体的功能仅在少数有限的情况下得到研究。如今,RNA测序技术为在全基因组范围内以无偏见的方式研究可变剪接提供了绝佳机会。随着公共数据库中数据的快速积累,有效整合许多不同的RNA测序数据集以研究可变剪接的迫切需求带来了新的挑战。本文讨论了一组先进的计算方法,这些方法可以整合和分析许多RNA测序数据集,以系统地识别剪接模块,揭示转录和剪接的耦合,并在全基因组范围内预测剪接异构体的功能。