Department of Computer Science, University of California, Berkeley, Berkeley, California, USA.
Nat Methods. 2013 Jan;10(1):71-3. doi: 10.1038/nmeth.2251. Epub 2012 Nov 18.
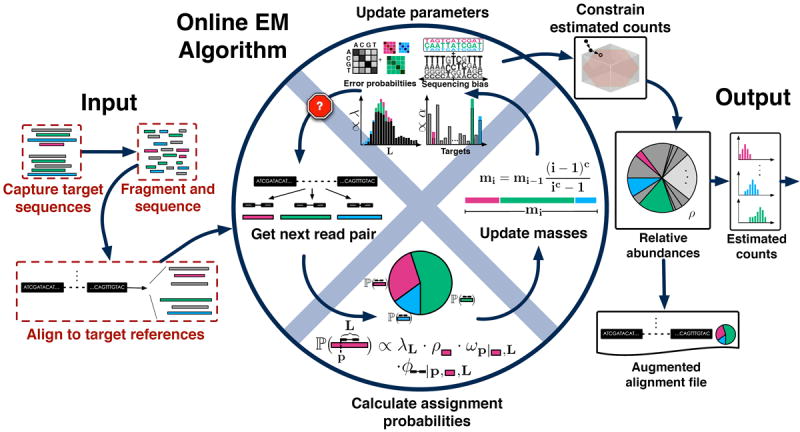
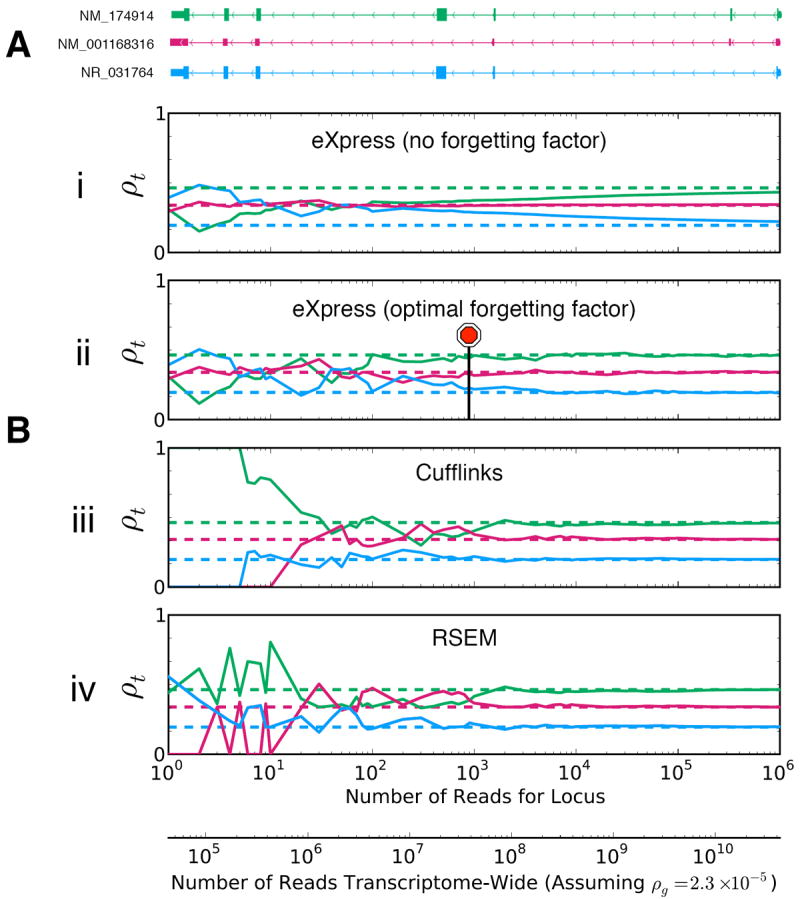
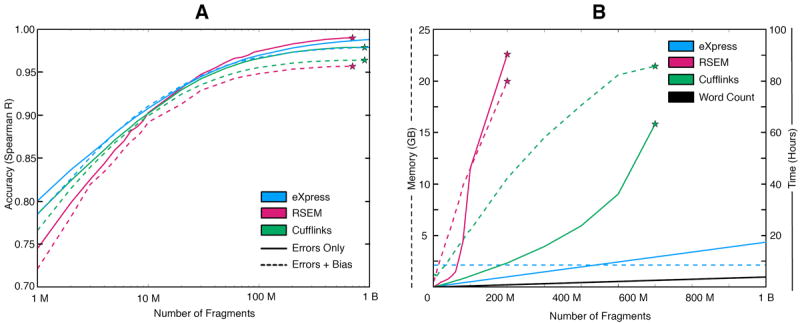
We present eXpress, a software package for efficient probabilistic assignment of ambiguously mapping sequenced fragments. eXpress uses a streaming algorithm with linear run time and constant memory use. It can determine abundances of sequenced molecules in real time and can be applied to ChIP-seq, metagenomics and other large-scale sequencing data. We demonstrate its use on RNA-seq data and show that eXpress achieves greater efficiency than other quantification methods.
我们介绍了 eXpress,这是一个用于高效概率分配模糊映射测序片段的软件包。eXpress 使用具有线性运行时间和恒定内存使用的流算法。它可以实时确定测序分子的丰度,并可应用于 ChIP-seq、宏基因组学和其他大规模测序数据。我们在 RNA-seq 数据上演示了它的使用,并表明 eXpress 比其他定量方法更有效率。