Tuggle Katherine L, Birket Susan E, Cui Xiaoxia, Hong Jeong, Warren Joe, Reid Lara, Chambers Andre, Ji Diana, Gamber Kevin, Chu Kengyeh K, Tearney Guillermo, Tang Li Ping, Fortenberry James A, Du Ming, Cadillac Joan M, Bedwell David M, Rowe Steven M, Sorscher Eric J, Fanucchi Michelle V
Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, Alabama, United States of America; Department of Environmental Health Sciences, School of Public Health, University of Alabama at Birmingham, Birmingham, Alabama, United States of America.
Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, Alabama, United States of America; Department of Medicine, University of Alabama at Birmingham, Birmingham, Alabama, United States of America.
PLoS One. 2014 Mar 7;9(3):e91253. doi: 10.1371/journal.pone.0091253. eCollection 2014.
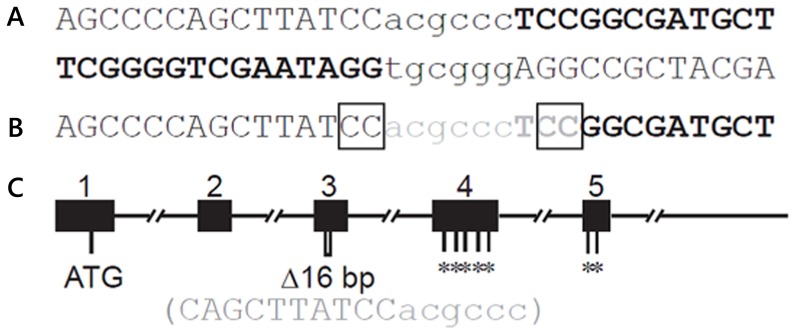
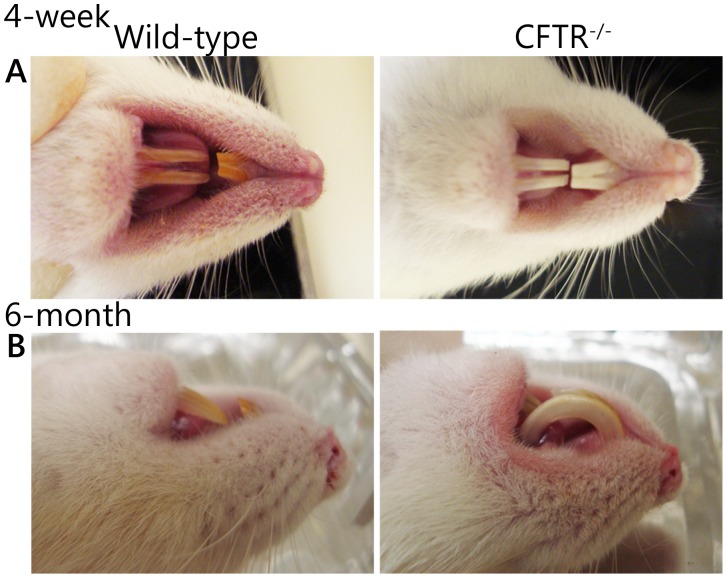
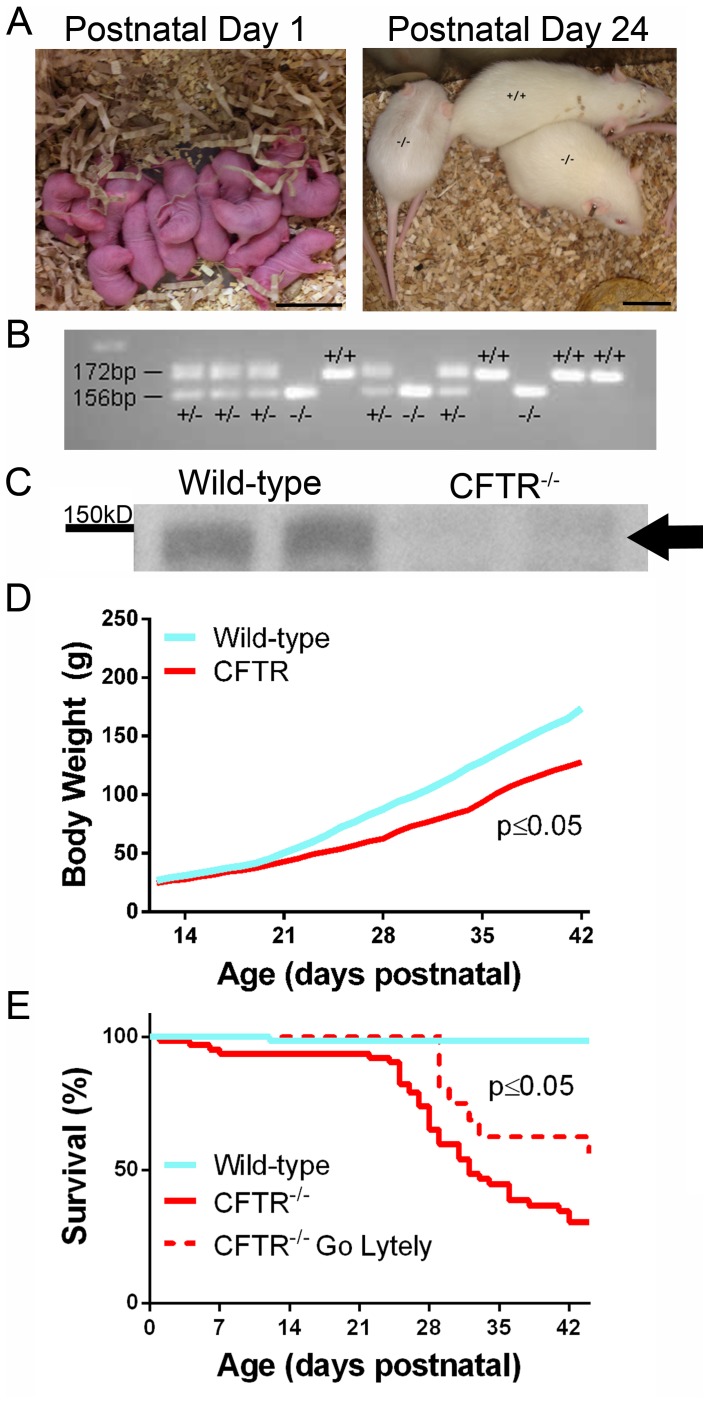
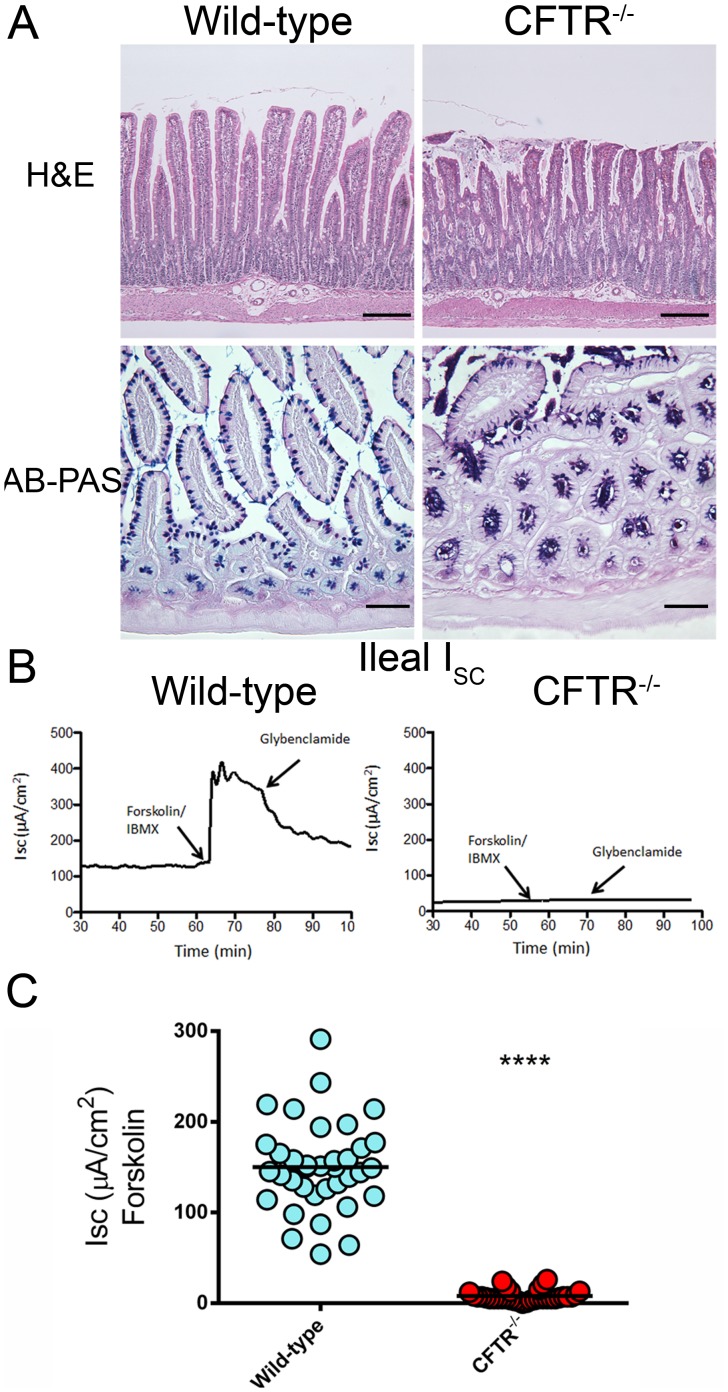
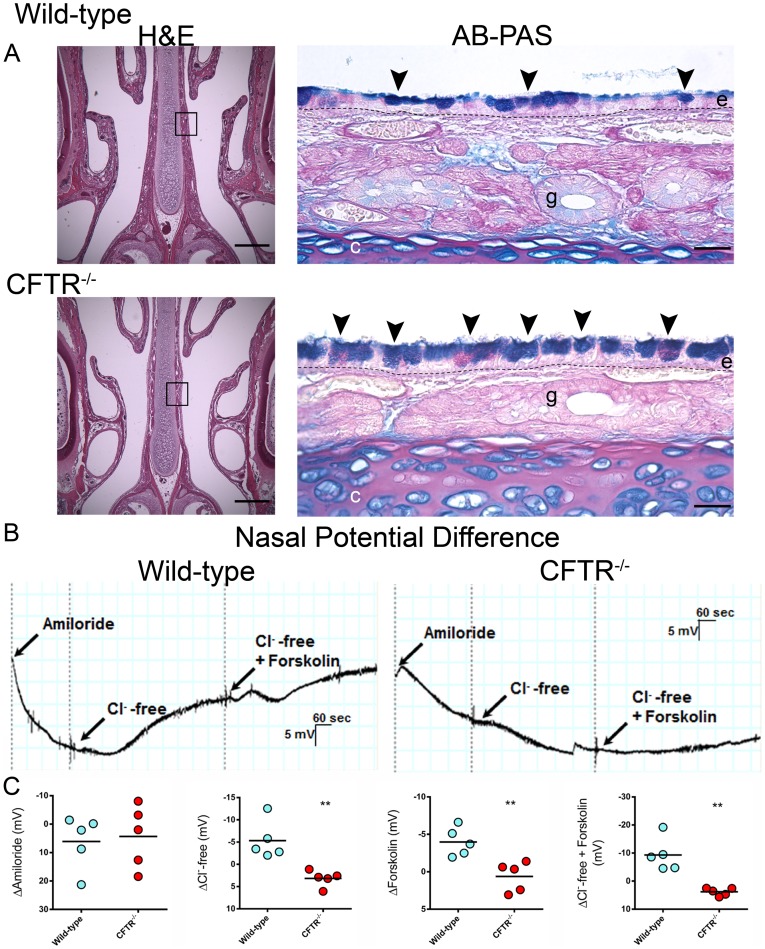
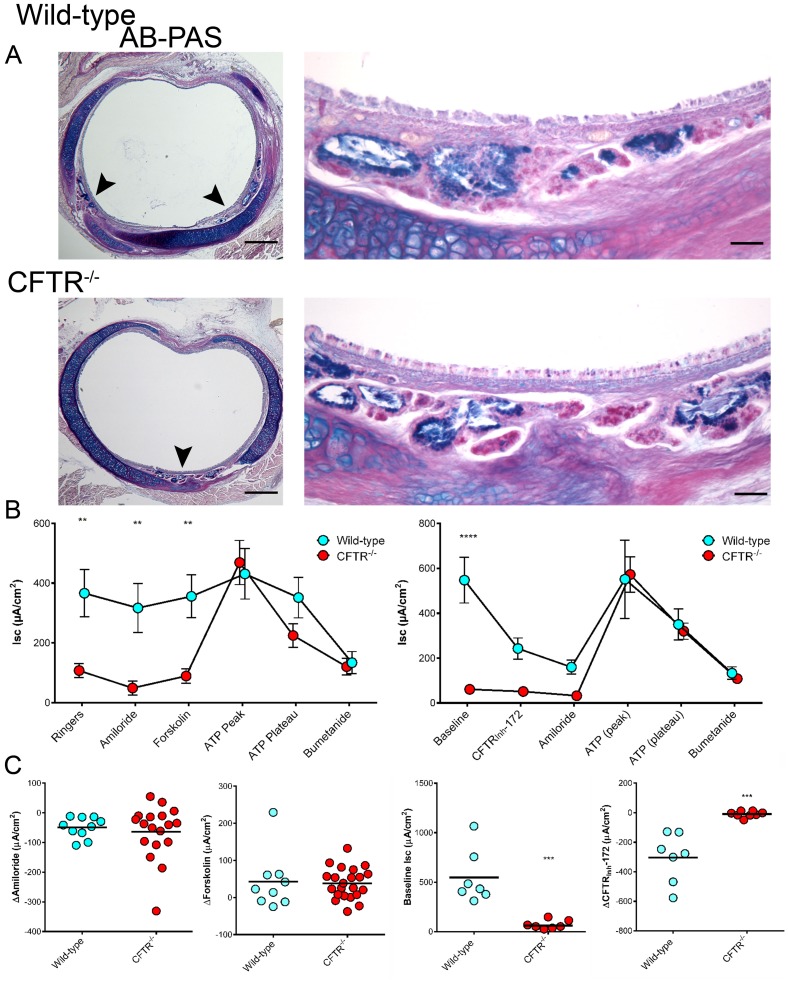
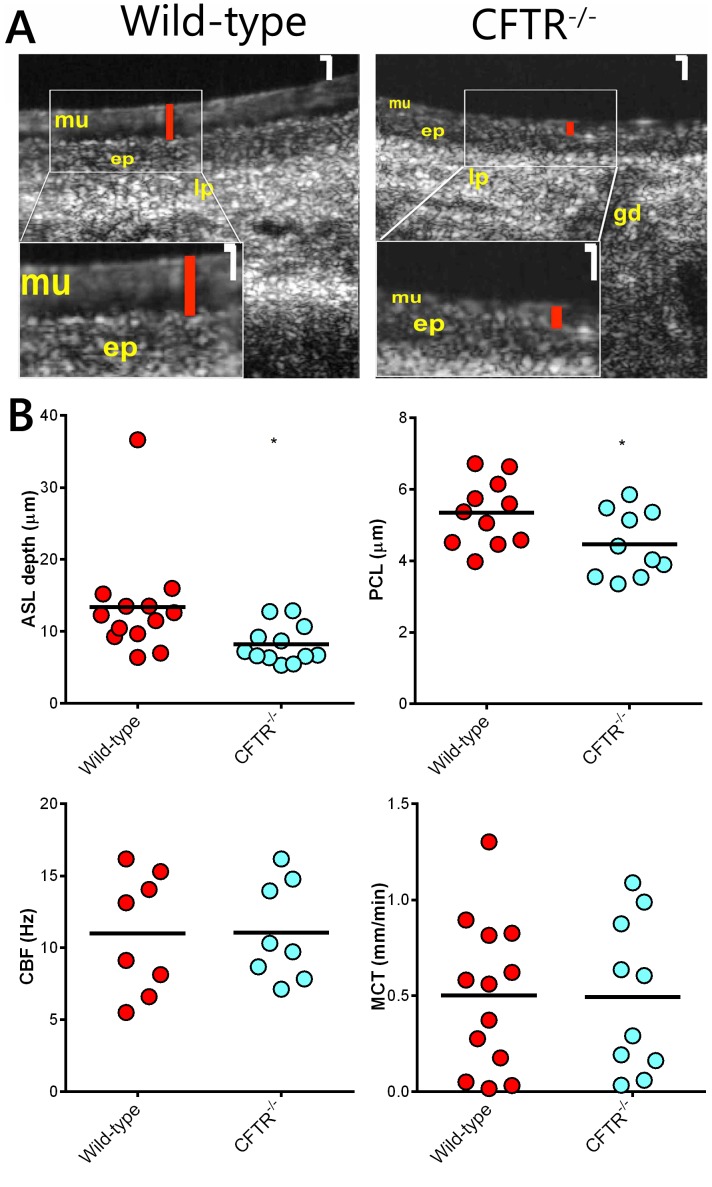
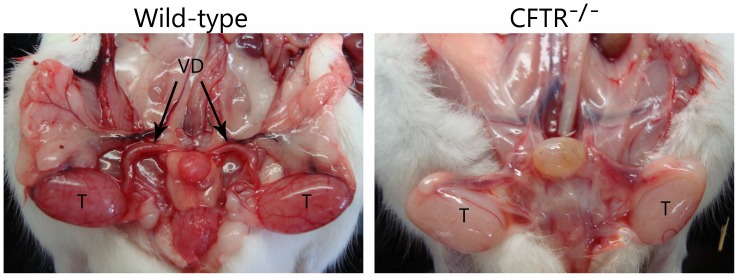
Animal models for cystic fibrosis (CF) have contributed significantly to our understanding of disease pathogenesis. Here we describe development and characterization of the first cystic fibrosis rat, in which the cystic fibrosis transmembrane conductance regulator gene (CFTR) was knocked out using a pair of zinc finger endonucleases (ZFN). The disrupted Cftr gene carries a 16 base pair deletion in exon 3, resulting in loss of CFTR protein expression. Breeding of heterozygous (CFTR+/-) rats resulted in Mendelian distribution of wild-type, heterozygous, and homozygous (CFTR-/-) pups. Nasal potential difference and transepithelial short circuit current measurements established a robust CF bioelectric phenotype, similar in many respects to that seen in CF patients. Young CFTR-/- rats exhibited histological abnormalities in the ileum and increased intracellular mucus in the proximal nasal septa. By six weeks of age, CFTR-/- males lacked the vas deferens bilaterally. Airway surface liquid and periciliary liquid depth were reduced, and submucosal gland size was abnormal in CFTR-/- animals. Use of ZFN based gene disruption successfully generated a CF animal model that recapitulates many aspects of human disease, and may be useful for modeling other CF genotypes, including CFTR processing defects, premature truncation alleles, and channel gating abnormalities.
囊性纤维化(CF)动物模型对我们理解疾病发病机制有显著贡献。在此,我们描述了首例囊性纤维化大鼠的培育及特性,其中利用一对锌指核酸酶(ZFN)敲除了囊性纤维化跨膜传导调节基因(CFTR)。被破坏的Cftr基因在外显子3中有16个碱基对缺失,导致CFTR蛋白表达缺失。杂合子(CFTR+/-)大鼠的繁殖产生了野生型、杂合子和纯合子(CFTR-/-)幼崽的孟德尔式分布。鼻电位差和跨上皮短路电流测量确定了一种强大的CF生物电表型,在许多方面与CF患者所见相似。年轻的CFTR-/-大鼠在回肠表现出组织学异常,在鼻中隔近端细胞内黏液增加。到六周龄时,CFTR-/-雄性大鼠双侧缺乏输精管。CFTR-/-动物的气道表面液体和纤毛周围液体深度降低,黏膜下腺大小异常。基于ZFN的基因破坏的应用成功产生了一种CF动物模型,该模型概括了人类疾病的许多方面,可能有助于模拟其他CF基因型,包括CFTR加工缺陷、过早截断等位基因和通道门控异常。