Bailey Derek J, McDevitt Molly T, Westphall Michael S, Pagliarini David J, Coon Joshua J
Department of Chemistry, University of Wisconsin - Madison , 1101 Unviersity Avenue, Madison, Wisconsin 53706, United States.
J Proteome Res. 2014 Apr 4;13(4):2152-61. doi: 10.1021/pr401278j. Epub 2014 Mar 18.
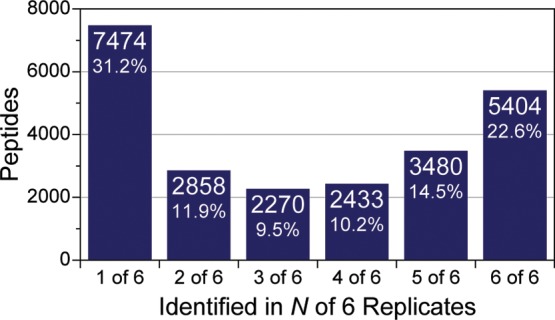
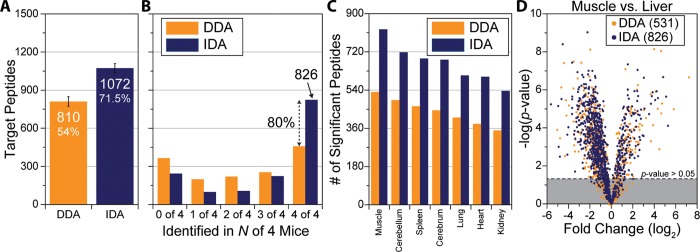
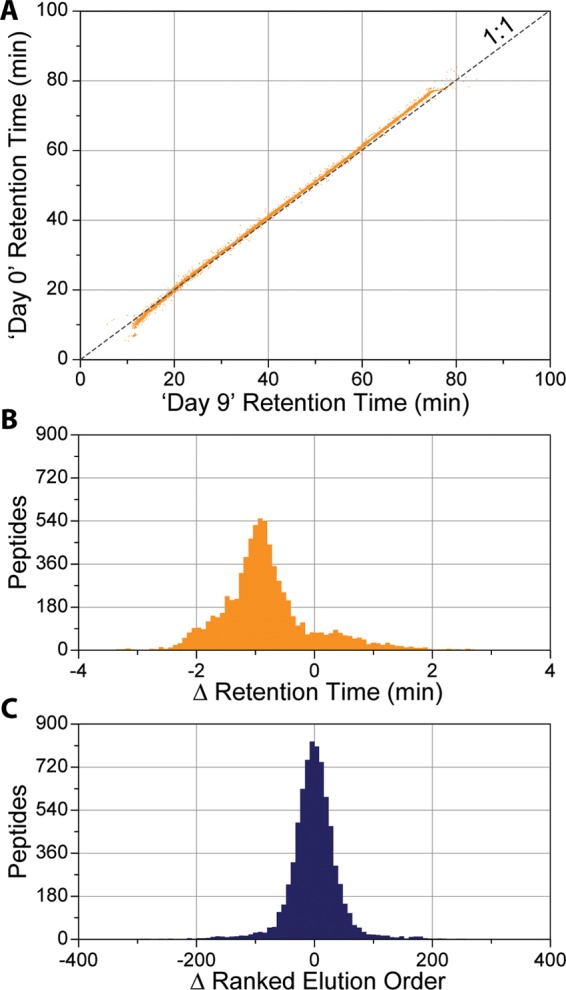
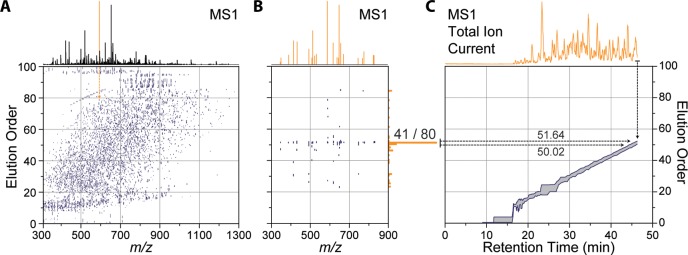
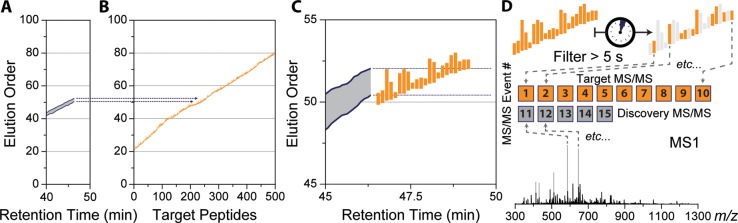
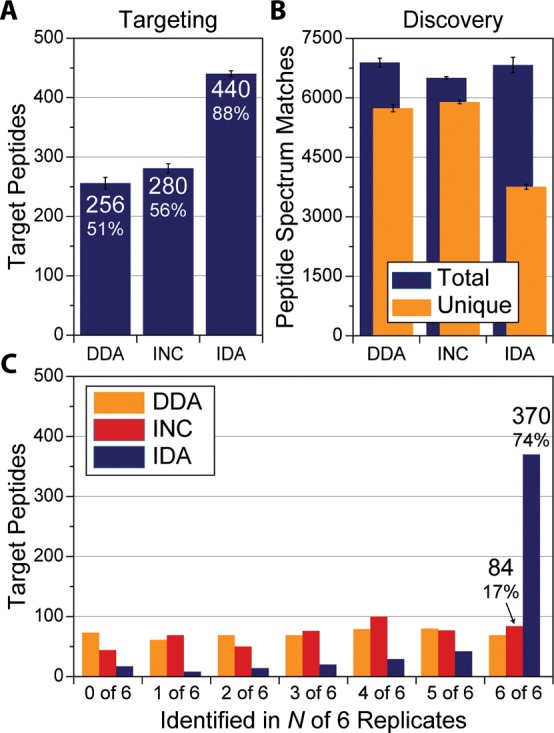
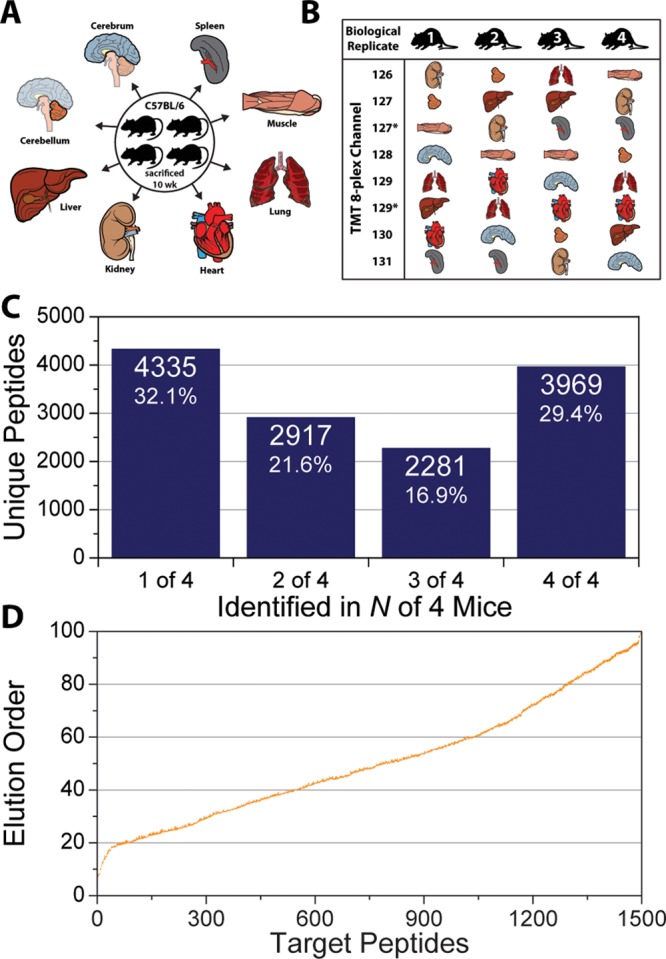
A mass spectrometry (MS) method is described here that can reproducibly identify hundreds of peptides across multiple experiments. The method uses intelligent data acquisition to precisely target peptides while simultaneously identifying thousands of other, nontargeted peptides in a single nano-LC-MS/MS experiment. We introduce an online peptide elution order alignment algorithm that targets peptides based on their relative elution order, eliminating the need for retention-time-based scheduling. We have applied this method to target 500 mouse peptides across six technical replicate nano-LC-MS/MS experiments and were able to identify 440 of these in all six, compared with only 256 peptides using data-dependent acquisition (DDA). A total of 3757 other peptides were also identified within the same experiment, illustrating that this hybrid method does not eliminate the novel discovery advantages of DDA. The method was also tested on a set of mice in biological quadruplicate and increased the number of identified target peptides in all four mice by over 80% (826 vs 459) compared with the standard DDA method. We envision real-time data analysis as a powerful tool to improve the quality and reproducibility of proteomic data sets.
本文描述了一种质谱(MS)方法,该方法能够在多个实验中可重复地鉴定数百种肽段。该方法使用智能数据采集来精确靶向肽段,同时在单次纳升液相色谱-串联质谱(nano-LC-MS/MS)实验中鉴定数千种其他非靶向肽段。我们引入了一种在线肽段洗脱顺序比对算法,该算法基于肽段的相对洗脱顺序来靶向肽段,从而无需基于保留时间的调度。我们已将此方法应用于在六个技术重复的nano-LC-MS/MS实验中靶向500种小鼠肽段,并且在所有六个实验中能够鉴定出其中的440种,相比之下,使用数据依赖型采集(DDA)仅能鉴定出256种肽段。在同一实验中还总共鉴定出了3757种其他肽段,这表明这种混合方法并未消除DDA在新发现方面的优势。该方法还在一组进行了生物学四重重复的小鼠上进行了测试,与标准DDA方法相比,所有四只小鼠中鉴定出的靶向肽段数量增加了80%以上(从459种增加到826种)。我们设想将实时数据分析作为提高蛋白质组数据集质量和可重复性的强大工具。