Computational and Systems Biology, Genome Institute of Singapore, 60 Biopolis Street, Singapore 138672, Singapore.
BMC Genomics. 2014 Mar 19;15:208. doi: 10.1186/1471-2164-15-208.
Cooperative binding of transcription factor (TF) dimers to DNA is increasingly recognized as a major contributor to binding specificity. However, it is likely that the set of known TF dimers is highly incomplete, given that they were discovered using ad hoc approaches, or through computational analyses of limited datasets.
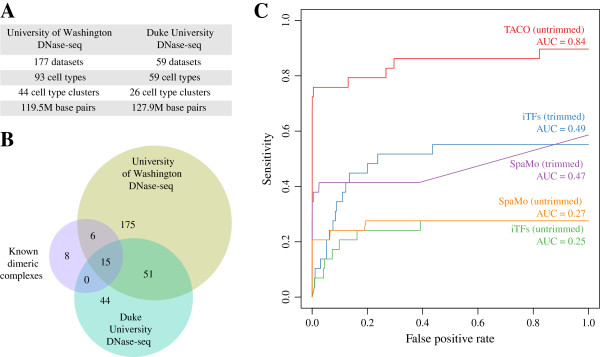
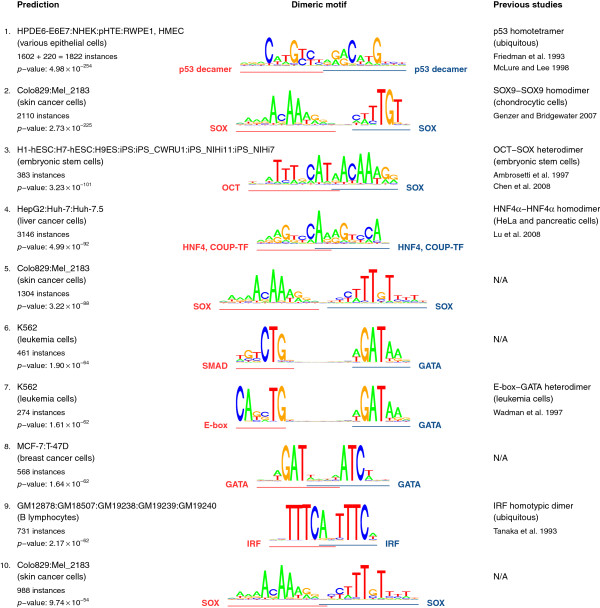
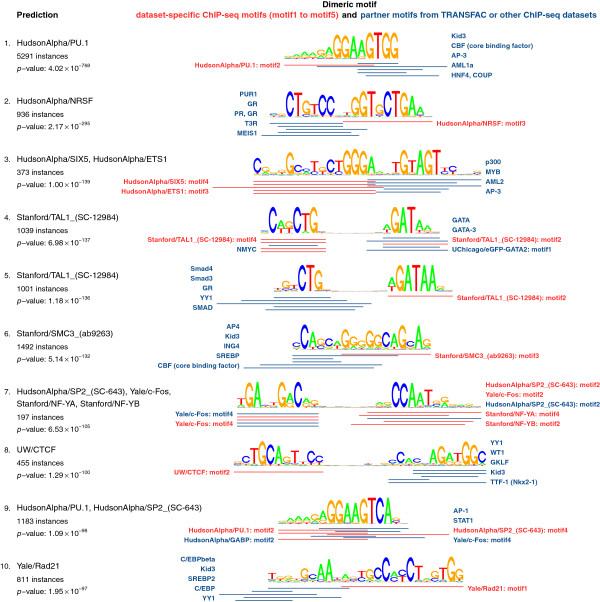
Here, we present TACO (Transcription factor Association from Complex Overrepresentation), a general-purpose standalone software tool that takes as input any genome-wide set of regulatory elements and predicts cell-type-specific TF dimers based on enrichment of motif complexes. TACO is the first tool that can accommodate motif complexes composed of overlapping motifs, a characteristic feature of many known TF dimers. Our method comprehensively outperforms existing tools when benchmarked on a reference set of 29 known dimers. We demonstrate the utility and consistency of TACO by applying it to 152 DNase-seq datasets and 94 ChIP-seq datasets.
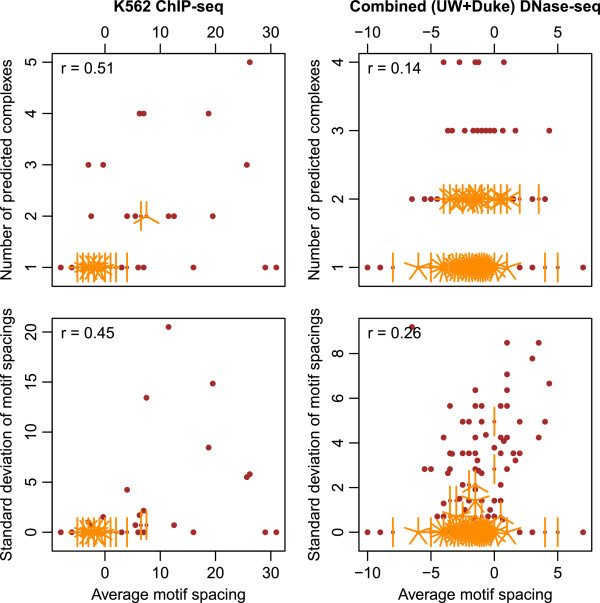
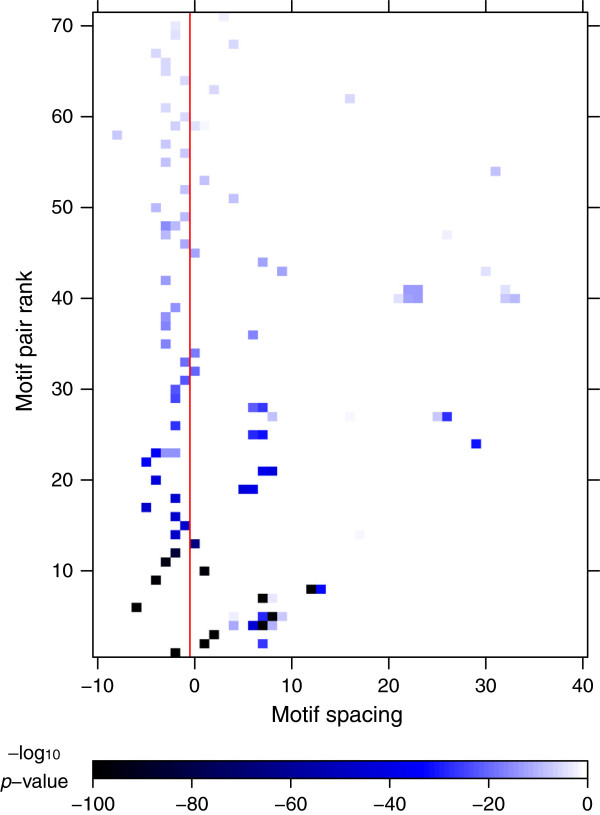
Based on these results, we uncover a general principle governing the structure of TF-TF-DNA ternary complexes, namely that the flexibility of the complex is correlated with, and most likely a consequence of, inter-motif spacing.
转录因子 (TF) 二聚体与 DNA 的协同结合作用越来越被认为是结合特异性的主要贡献因素。然而,鉴于它们是通过特定的方法或通过对有限数据集的计算分析发现的,因此已知的 TF 二聚体的集合很可能是高度不完全的。
在这里,我们提出了 TACO(转录因子复合物的关联),这是一个通用的独立软件工具,它可以接受任何全基因组的调控元件集作为输入,并基于基序复合物的富集来预测特定细胞类型的 TF 二聚体。TACO 是第一个可以容纳由重叠基序组成的基序复合物的工具,这是许多已知的 TF 二聚体的一个特征。当在 29 个已知二聚体的参考集上进行基准测试时,我们的方法在全面性能上优于现有工具。我们通过将其应用于 152 个 DNase-seq 数据集和 94 个 ChIP-seq 数据集,展示了 TACO 的实用性和一致性。
基于这些结果,我们揭示了一个普遍的原则,即 TF-TF-DNA 三元复合物的结构,即复合物的灵活性与基序之间的间隔有关,而且很可能是其结果。