Laboratory of Systems Immunology, Immunology Frontier Research Center, Osaka University, 3-1 Yamada-oka, Suita, Osaka 565-0871, Japan.
BMC Genomics. 2012;13 Suppl 7(Suppl 7):S11. doi: 10.1186/1471-2164-13-S7-S11. Epub 2012 Dec 13.
Multiple transcription factors (TFs) are involved in the generation of gene expression patterns, such as tissue-specific gene expression and pleiotropic immune responses. However, how combinations of TFs orchestrate diverse gene expression patterns is poorly understood. Here we propose a new measure for regulatory motif co-occurrence and a new methodology to systematically identify TF pairs significantly co-occurring in a set of promoter sequences.
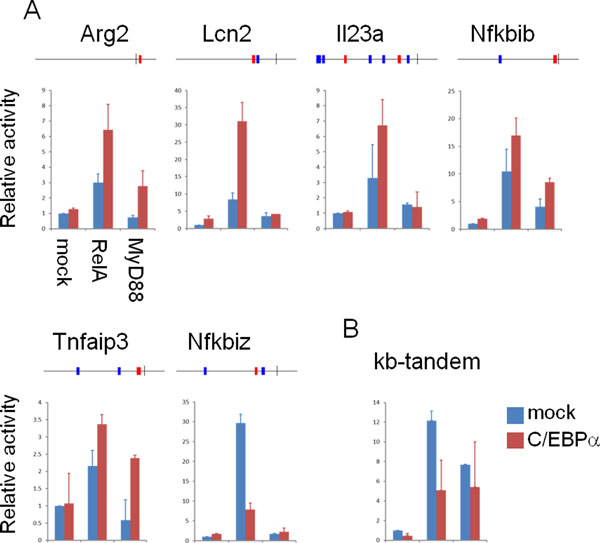
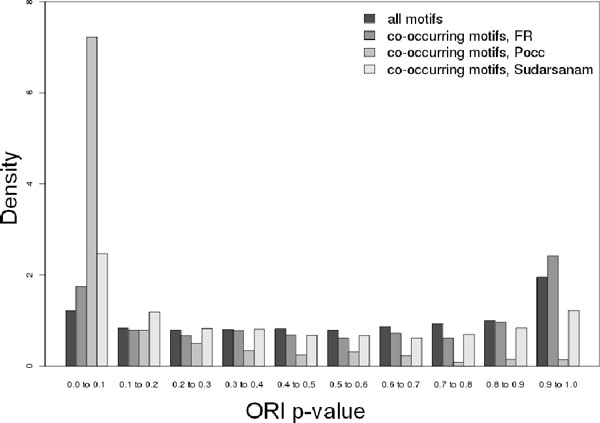
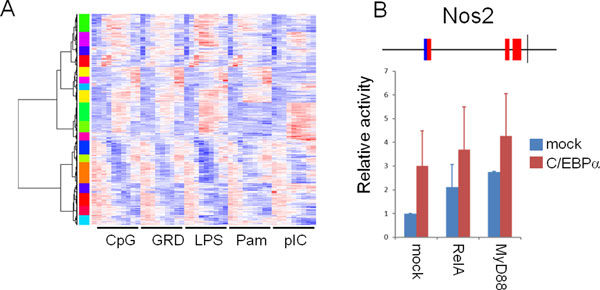
Initial analyses suggest that non-CpG promoters have a higher potential for combinatorial regulation than CpG island-associated promoters, and that co-occurrences are strongly influenced by motif similarity. We applied our method to large-scale gene expression data from various tissues, and showed how our measure for motif co-occurrence is not biased by motif over-representation. Our method identified, amongst others, the binding motifs of HNF1 and FOXP1 to be significantly co-occurring in promoters of liver/kidney specific genes. Binding sites tend to be positioned proximally to each other, suggesting interactions exist between this pair of transcription factors. Moreover, the binding sites of several TFs were found to co-occur with NF-κB and IRF sites in sets of genes with similar expression patterns in dendritic cells after Toll-like receptor stimulation. Of these, we experimentally verified that CCAAT enhancer binding protein alpha positively regulates its target promoters synergistically with NF-κB.
Both computational and experimental results indicate that the proposed method can clarify TF interactions that could not be observed by currently available prediction methods.
多个转录因子(TFs)参与基因表达模式的产生,例如组织特异性基因表达和多效性免疫反应。然而,TFs 组合如何协调不同的基因表达模式还知之甚少。在这里,我们提出了一种新的调控基序共现度量标准和一种系统识别启动子序列中显著共现的 TF 对的新方法。
初步分析表明,非 CpG 启动子比 CpG 岛相关启动子具有更高的组合调控潜力,而且共现受基序相似性的强烈影响。我们将我们的方法应用于来自各种组织的大规模基因表达数据,并展示了我们的基序共现度量标准如何不受基序过表达的影响。我们的方法鉴定了 HNF1 和 FOXP1 的结合基序在肝脏/肾脏特异性基因的启动子中显著共现,除此之外还有其他的。结合位点往往彼此靠近定位,表明这对转录因子之间存在相互作用。此外,在 Toll 样受体刺激后树突状细胞中具有相似表达模式的基因集中,发现几个 TF 的结合位点与 NF-κB 和 IRF 位点共现。在这些基因中,我们通过实验验证了 CCAAT 增强子结合蛋白α与 NF-κB 协同正向调节其靶启动子。
计算和实验结果均表明,所提出的方法可以阐明当前可用的预测方法无法观察到的 TF 相互作用。