Key Laboratory of Animal Genetics and Breeding of Ministry of Agriculture, National Engineering Laboratory of Animal Breeding, College of Animal Science and Technology, China Agricultural University, Beijing 100193, China.
BMC Genomics. 2014 Mar 24;15:226. doi: 10.1186/1471-2164-15-226.
Recently, RNA sequencing (RNA-seq) has rapidly emerged as a major transcriptome profiling system. Elucidation of the bovine mammary gland transcriptome by RNA-seq is essential for identifying candidate genes that contribute to milk composition traits in dairy cattle.
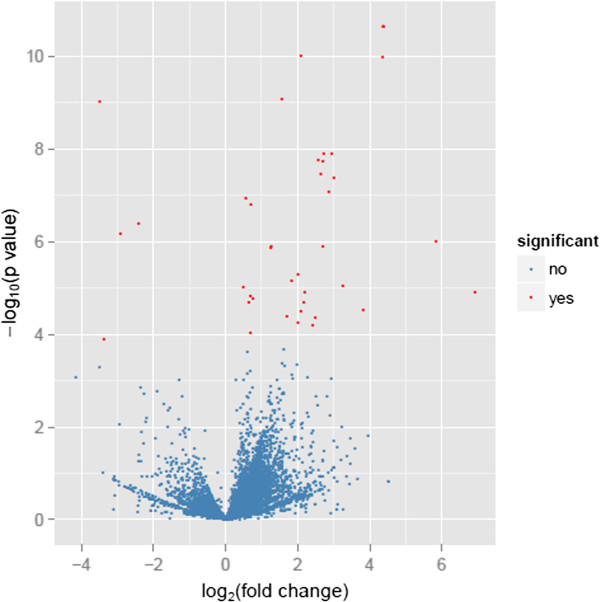
We used massive, parallel, high-throughput, RNA-seq to generate the bovine transcriptome from the mammary glands of four lactating Holstein cows with extremely high and low phenotypic values of milk protein and fat percentage. In total, we obtained 48,967,376-75,572,578 uniquely mapped reads that covered 82.25% of the current annotated transcripts, which represented 15549 mRNA transcripts, across all the four mammary gland samples. Among them, 31 differentially expressed genes (p < 0.05, false discovery rate q < 0.05) between the high and low groups of cows were revealed. Gene ontology and pathway analysis demonstrated that the 31 differently expressed genes were enriched in specific biological processes with regard to protein metabolism, fat metabolism, and mammary gland development (p < 0.05). Integrated analysis of differential gene expression, previously reported quantitative trait loci, and genome-wide association studies indicated that TRIB3, SAA (SAA1, SAA3, and M-SAA3.2), VEGFA, PTHLH, and RPL23A were the most promising candidate genes affecting milk protein and fat percentage.
This study investigated the complexity of the mammary gland transcriptome in dairy cattle using RNA-seq. Integrated analysis of differential gene expression and the reported quantitative trait loci and genome-wide association study data permitted the identification of candidate key genes for milk composition traits.
最近,RNA 测序(RNA-seq)迅速成为一种主要的转录组分析系统。阐明牛乳腺转录组对于鉴定与奶牛乳成分性状相关的候选基因至关重要。
我们使用大规模、平行、高通量的 RNA-seq 技术,从 4 头泌乳荷斯坦奶牛的乳腺中生成了转录组,这些奶牛的乳蛋白和乳脂率表型值极高和极低。总共获得了 48967376-75572578 个唯一映射的读数,覆盖了当前注释转录本的 82.25%,代表了所有 4 个乳腺样本中的 15549 个 mRNA 转录本。其中,在高组和低组奶牛之间揭示了 31 个差异表达基因(p<0.05,错误发现率 q<0.05)。基因本体论和途径分析表明,31 个差异表达基因在蛋白质代谢、脂肪代谢和乳腺发育方面富集了特定的生物学过程(p<0.05)。差异基因表达、先前报道的数量性状位点和全基因组关联研究的综合分析表明,TRIB3、SAA(SAA1、SAA3 和 M-SAA3.2)、VEGFA、PTHLH 和 RPL23A 是影响乳蛋白和乳脂率的最有希望的候选基因。
本研究使用 RNA-seq 研究了奶牛乳腺转录组的复杂性。差异基因表达和报道的数量性状位点以及全基因组关联研究数据的综合分析允许鉴定乳成分性状的候选关键基因。