Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health Bethesda, MD, USA.
Front Genet. 2014 Mar 18;5:53. doi: 10.3389/fgene.2014.00053. eCollection 2014.
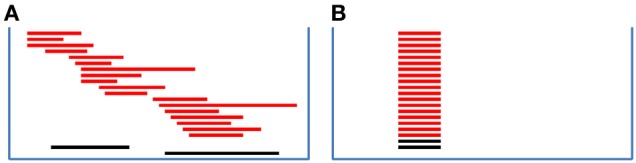
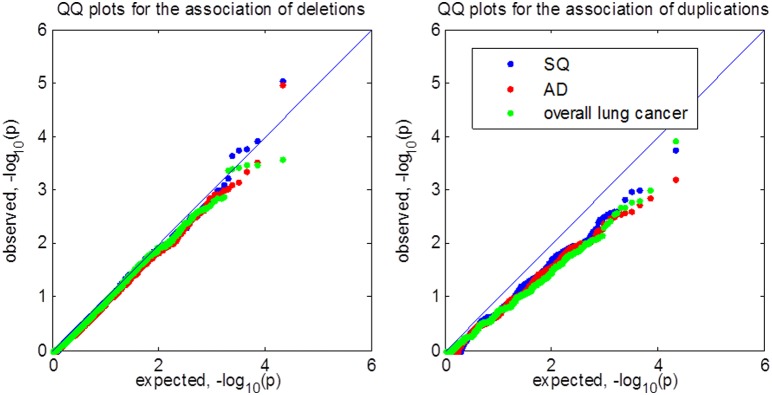
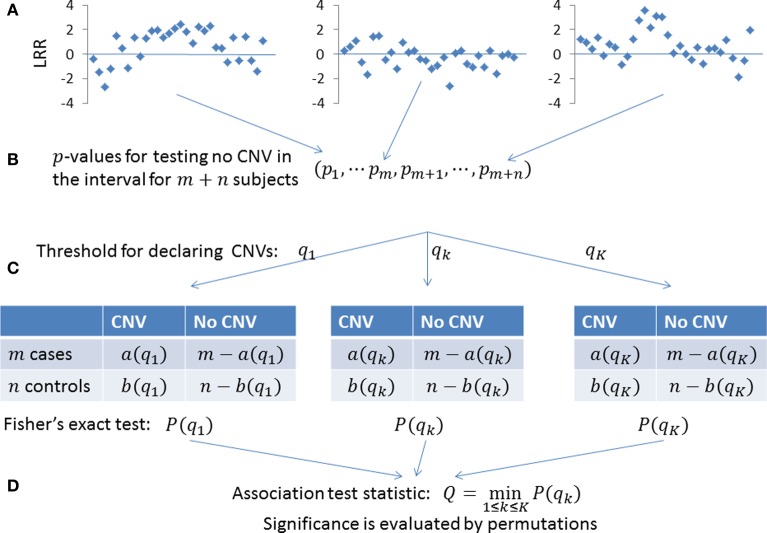
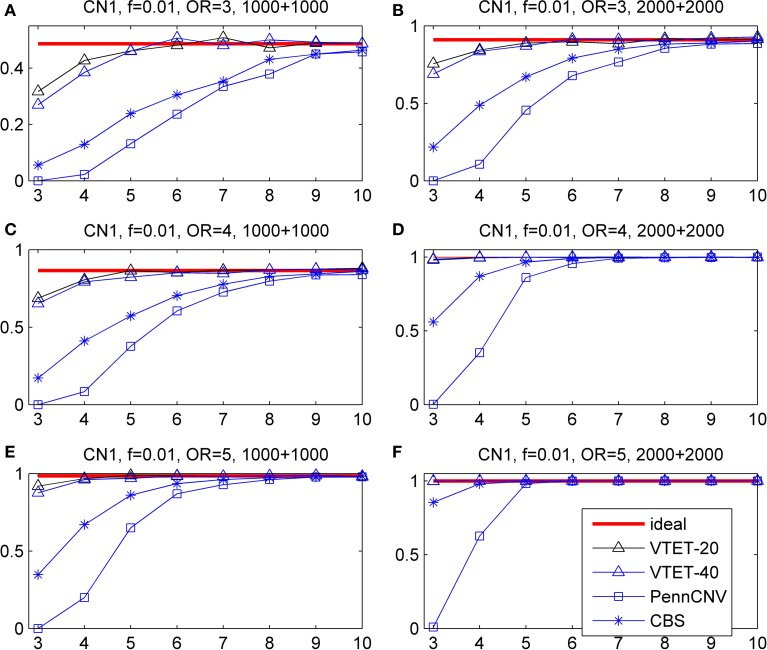
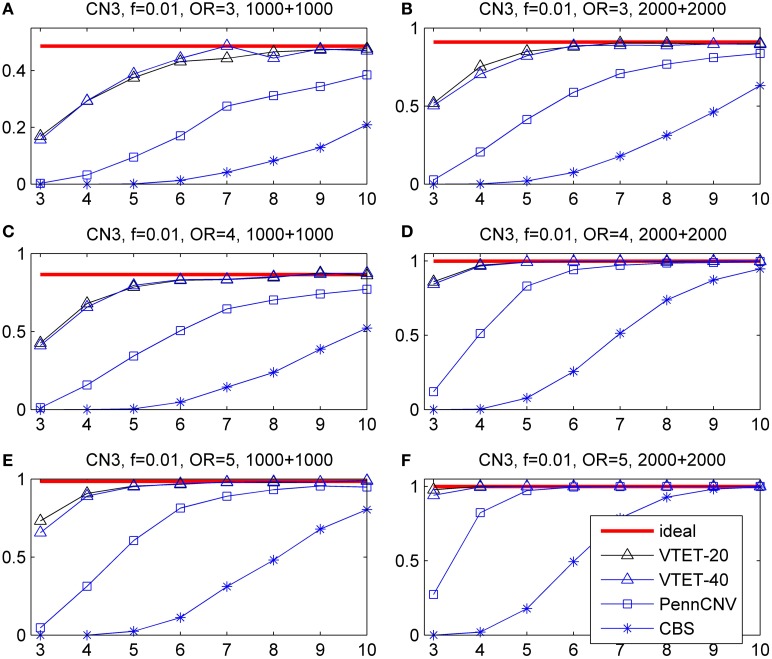
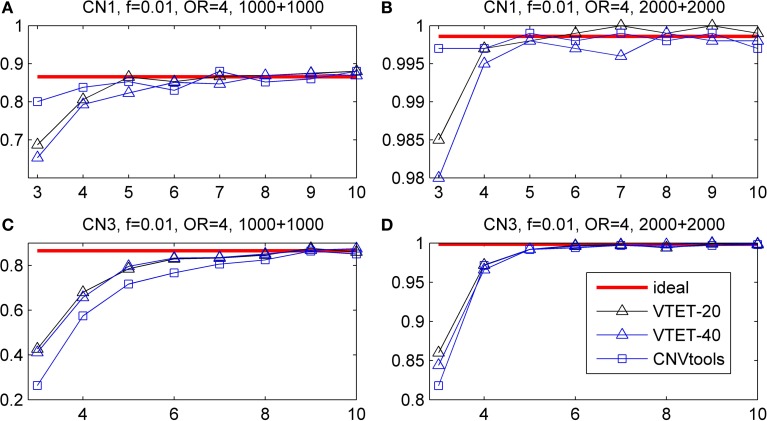
Copy number variations (CNVs) constitute a major source of genetic variations in human populations and have been reported to be associated with complex diseases. Methods have been developed for detecting CNVs and testing CNV associations in genome-wide association studies (GWAS) based on SNP arrays. Commonly used two-step testing procedures work well only for long CNVs while direct CNV association testing methods work only for recurrent CNVs. Assuming that short CNVs disrupting any part of a given genomic region increase disease risk, we developed a variable threshold exact test (VTET) for testing disease associations of CNVs randomly distributed in the genome using intensity data from SNP arrays. By extensive simulations, we found that VTET outperformed two-step testing procedures based on existing CNV calling algorithms for short CNVs and that the performance of VTET was robust to the length of the genomic region. In addition, VTET had a comparable performance with CNVtools for testing the association of recurrent CNVs. Thus, we expect VTET to be useful for testing disease associations of both recurrent and randomly distributed CNVs using existing GWAS data. We applied VTET to a lung cancer GWAS and identified a genome-wide significant region on chromosome 18q22.3 for lung squamous cell carcinoma.
拷贝数变异 (CNVs) 是人类群体中遗传变异的主要来源,已有报道称其与复杂疾病有关。已经开发出了用于检测 CNVs 和在全基因组关联研究 (GWAS) 中检测 CNV 关联的方法,这些方法基于 SNP 阵列。常用的两步测试程序仅适用于长 CNVs,而直接 CNV 关联测试方法仅适用于重复 CNVs。假设短 CNVs 破坏给定基因组区域的任何部分都会增加疾病风险,我们开发了一种可变阈值精确检验 (VTET),用于使用 SNP 阵列的强度数据测试随机分布在基因组中的 CNV 与疾病的关联。通过广泛的模拟,我们发现 VTET 优于基于现有 CNV 调用算法的两步测试程序,用于短 CNVs,并且 VTET 的性能对基因组区域的长度具有鲁棒性。此外,VTET 在测试重复 CNV 的关联方面与 CNVtools 具有可比的性能。因此,我们预计 VTET 将有助于使用现有的 GWAS 数据测试重复和随机分布的 CNV 与疾病的关联。我们将 VTET 应用于肺癌 GWAS,并在 18q22.3 染色体上鉴定出一个与肺鳞状细胞癌相关的全基因组显著区域。