Department of Statistics, University of California, Los Angeles, CA, USA.
BMC Bioinformatics. 2012 Aug 16;13:205. doi: 10.1186/1471-2105-13-205.
Variations in DNA copy number carry information on the modalities of genome evolution and mis-regulation of DNA replication in cancer cells. Their study can help localize tumor suppressor genes, distinguish different populations of cancerous cells, and identify genomic variations responsible for disease phenotypes. A number of different high throughput technologies can be used to identify copy number variable sites, and the literature documents multiple effective algorithms. We focus here on the specific problem of detecting regions where variation in copy number is relatively common in the sample at hand. This problem encompasses the cases of copy number polymorphisms, related samples, technical replicates, and cancerous sub-populations from the same individual.
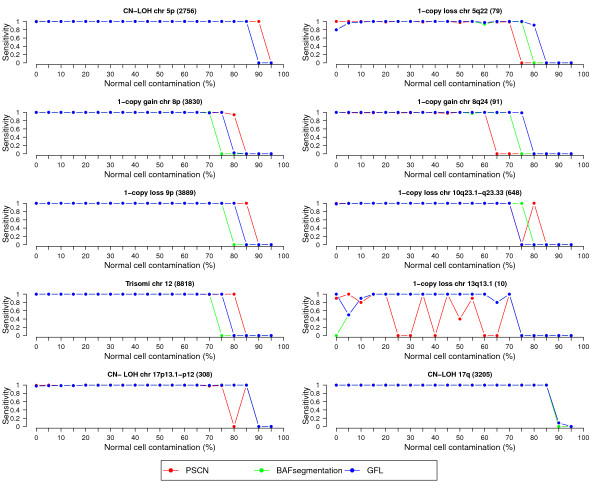
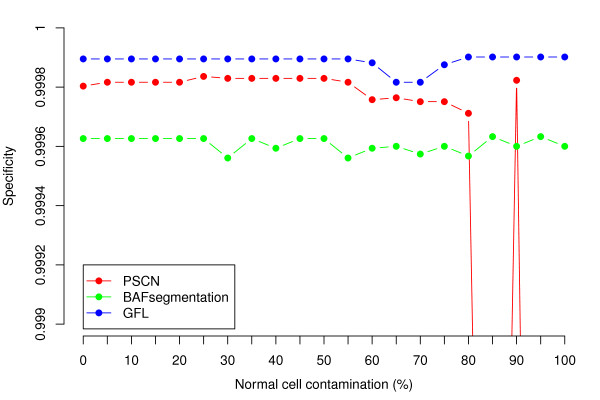
We present a segmentation method named generalized fused lasso (GFL) to reconstruct copy number variant regions. GFL is based on penalized estimation and is capable of processing multiple signals jointly. Our approach is computationally very attractive and leads to sensitivity and specificity levels comparable to those of state-of-the-art specialized methodologies. We illustrate its applicability with simulated and real data sets.
The flexibility of our framework makes it applicable to data obtained with a wide range of technology. Its versatility and speed make GFL particularly useful in the initial screening stages of large data sets.
DNA 拷贝数的变化携带了基因组进化和癌细胞中 DNA 复制失调的方式信息。对其进行研究有助于定位肿瘤抑制基因,区分癌细胞的不同群体,并识别导致疾病表型的基因组变异。有许多不同的高通量技术可用于识别拷贝数可变位点,并且文献中记录了多种有效的算法。我们在这里关注的是在手头样本中检测拷贝数变化相对常见的区域的具体问题。这个问题包括拷贝数多态性、相关样本、技术重复和来自同一个体的癌症亚群的情况。
我们提出了一种名为广义融合套索(GFL)的分割方法,用于重建拷贝数变异区域。GFL 基于惩罚估计,能够联合处理多个信号。我们的方法在计算上非常有吸引力,并且可以达到与最先进的专业方法相当的灵敏度和特异性水平。我们使用模拟和真实数据集来说明其适用性。
我们的框架的灵活性使其适用于各种技术获得的数据。其通用性和速度使得 GFL 在大型数据集的初始筛选阶段特别有用。