Ping Zheng, Siegal Gene P, Almeida Jonas S, Schnitt Stuart J, Shen Dejun
Department of Pathology, Division of Anatomic Pathology, Comprehensive Cancer Center, University of Alabama at Birmingham, Birmingham, Alabama, USA.
Department of Pathology, Division of Informatics, Comprehensive Cancer Center, University of Alabama at Birmingham, Birmingham, Alabama, USA.
J Pathol Inform. 2014 Jan 31;5(1):3. doi: 10.4103/2153-3539.126147. eCollection 2014.
Genetics and genomics have radically altered our understanding of breast cancer progression. However, the genomic basis of various histopathologic features of breast cancer is not yet well-defined.
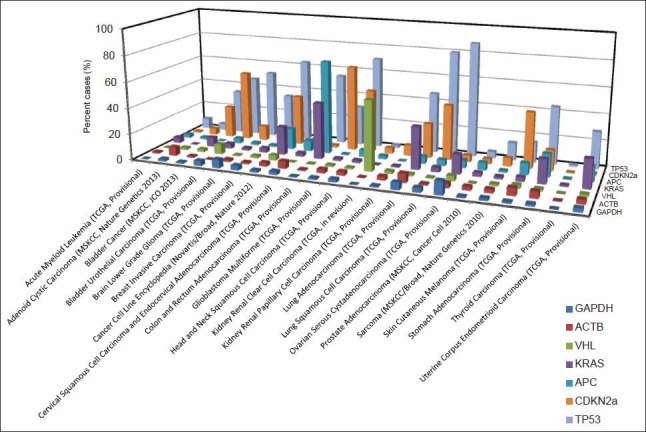
The Cancer Genome Atlas (TCGA) is an international database containing a large collection of human cancer genome sequencing data. cBioPortal is a web tool developed for mining these sequencing data. We performed mining of TCGA sequencing data in an attempt to characterize the genomic features correlated with breast cancer histopathology. We first assessed the quality of the TCGA data using a group of genes with known alterations in various cancers. Both genome-wide gene mutation and copy number changes as well as a group of genes with a high frequency of genetic changes were then correlated with various histopathologic features of invasive breast cancer.
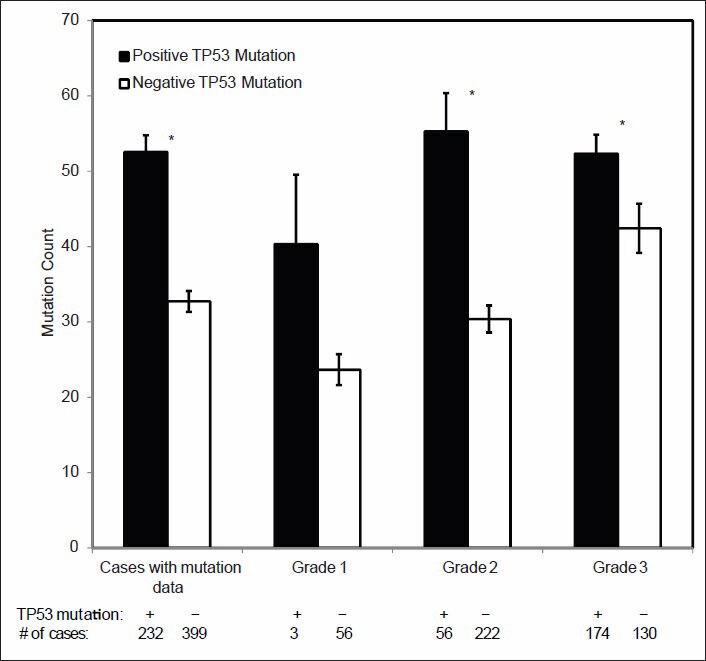
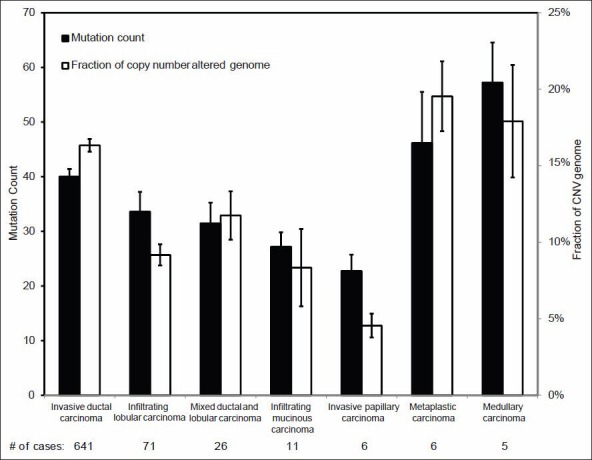
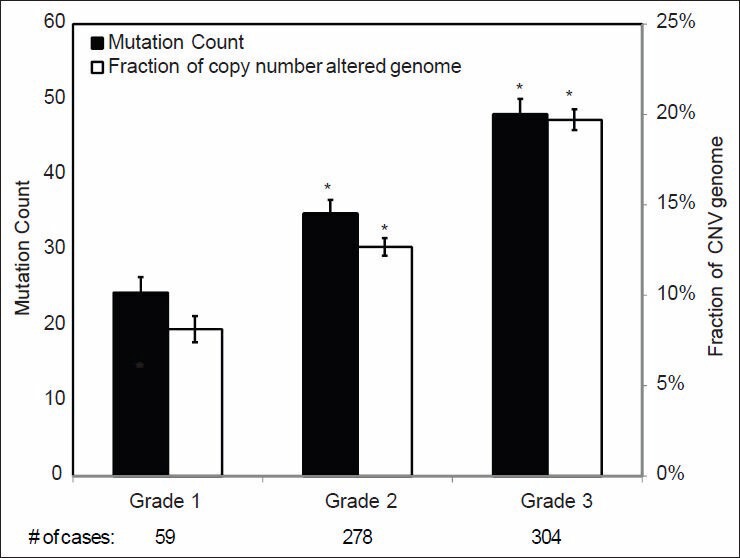
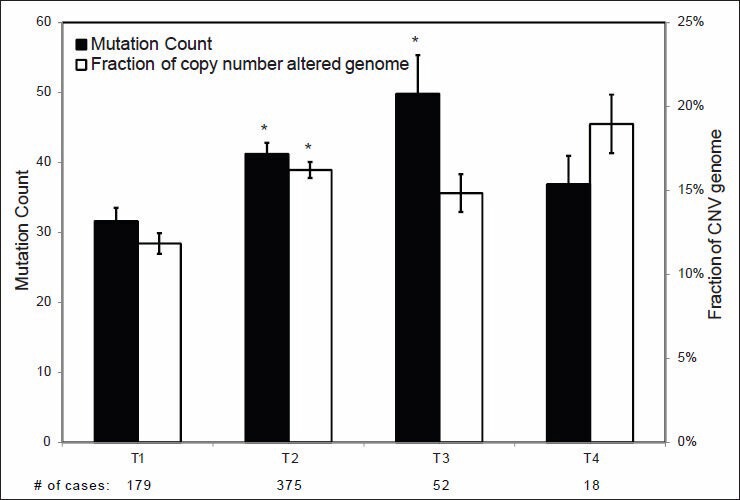
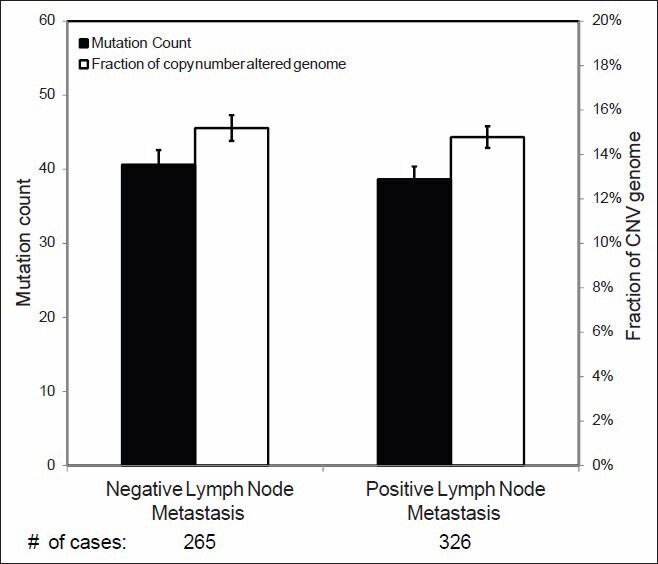
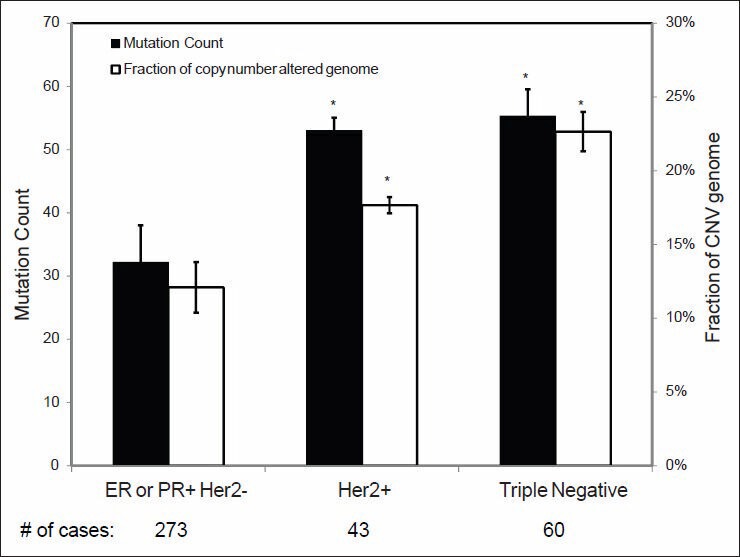
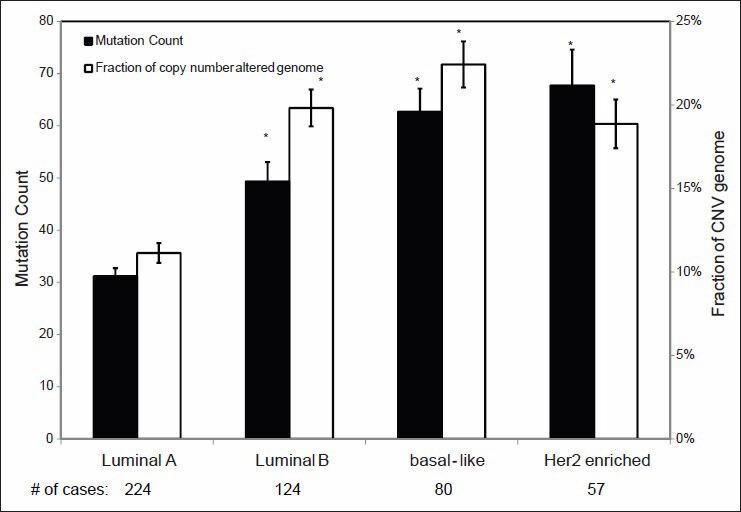
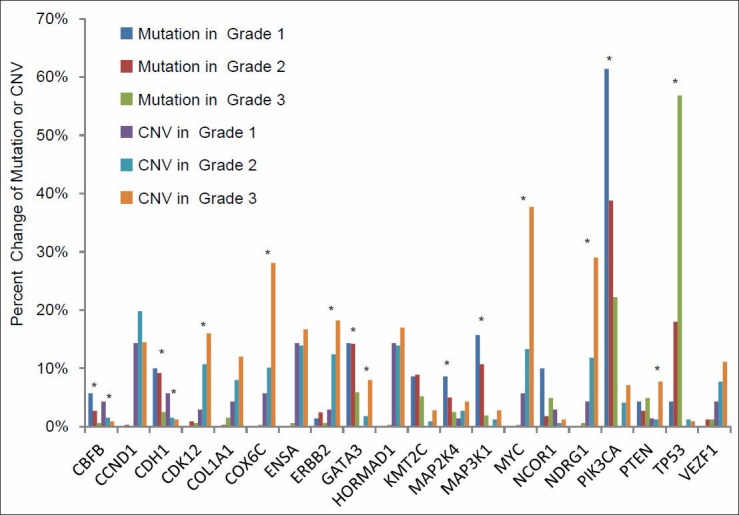
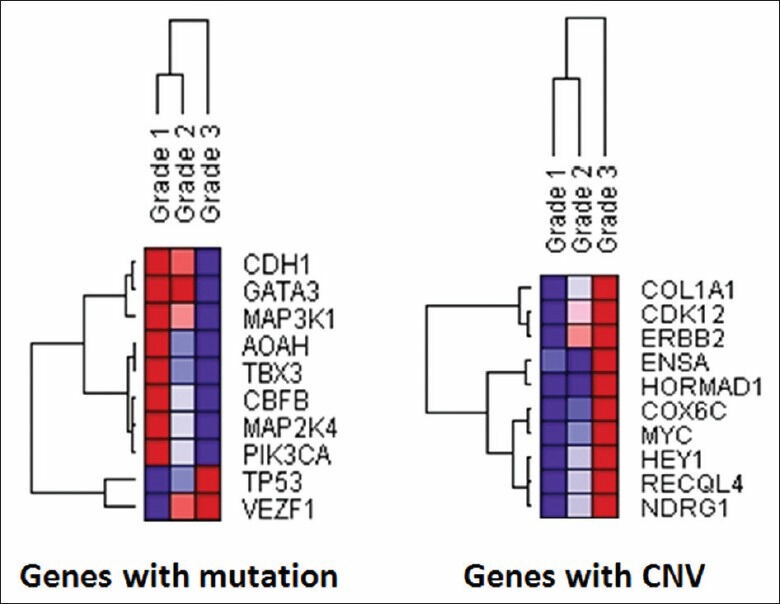
Validation of TCGA data using a group of genes with known alterations in breast cancer suggests that the TCGA has accurately documented the genomic abnormalities of multiple malignancies. Further analysis of TCGA breast cancer sequencing data shows that accumulation of specific genomic defects is associated with higher tumor grade, larger tumor size and receptor negativity. Distinct groups of genomic changes were found to be associated with the different grades of invasive ductal carcinoma. The mutator role of the TP53 gene was validated by genomic sequencing data of invasive breast cancer and TP53 mutation was found to play a critical role in defining high tumor grade.
Data mining of the TCGA genome sequencing data is an innovative and reliable method to help characterize the genomic abnormalities associated with histopathologic features of invasive breast cancer.
遗传学和基因组学已彻底改变了我们对乳腺癌进展的理解。然而,乳腺癌各种组织病理学特征的基因组基础尚未明确界定。
癌症基因组图谱(TCGA)是一个国际数据库,包含大量人类癌症基因组测序数据。cBioPortal是一个为挖掘这些测序数据而开发的网络工具。我们对TCGA测序数据进行挖掘,试图描绘与乳腺癌组织病理学相关的基因组特征。我们首先使用一组在各种癌症中具有已知改变的基因评估TCGA数据的质量。然后将全基因组基因突变和拷贝数变化以及一组具有高频率基因变化的基因与浸润性乳腺癌的各种组织病理学特征相关联。
使用一组在乳腺癌中具有已知改变的基因对TCGA数据进行验证表明,TCGA已准确记录了多种恶性肿瘤的基因组异常。对TCGA乳腺癌测序数据的进一步分析表明,特定基因组缺陷的积累与更高的肿瘤分级、更大的肿瘤大小和受体阴性相关。发现不同组的基因组变化与不同分级的浸润性导管癌相关。TP53基因的突变作用通过浸润性乳腺癌的基因组测序数据得到验证,并且发现TP53突变在定义高肿瘤分级中起关键作用。
对TCGA基因组测序数据进行数据挖掘是一种创新且可靠的方法,有助于描绘与浸润性乳腺癌组织病理学特征相关的基因组异常。