Brar Manreetpal Singh, Shi Mang, Hui Raymond Kin-Hi, Leung Frederick Chi-Ching
School of Biological Sciences, The University of Hong Kong, Hong Kong, China.
Sydney Emerging Infections & Biosecurity Institute, School of Biological Sciences and Sydney Medical School, The University of Sydney, Darlington, Australia.
PLoS One. 2014 Apr 3;9(4):e88807. doi: 10.1371/journal.pone.0088807. eCollection 2014.
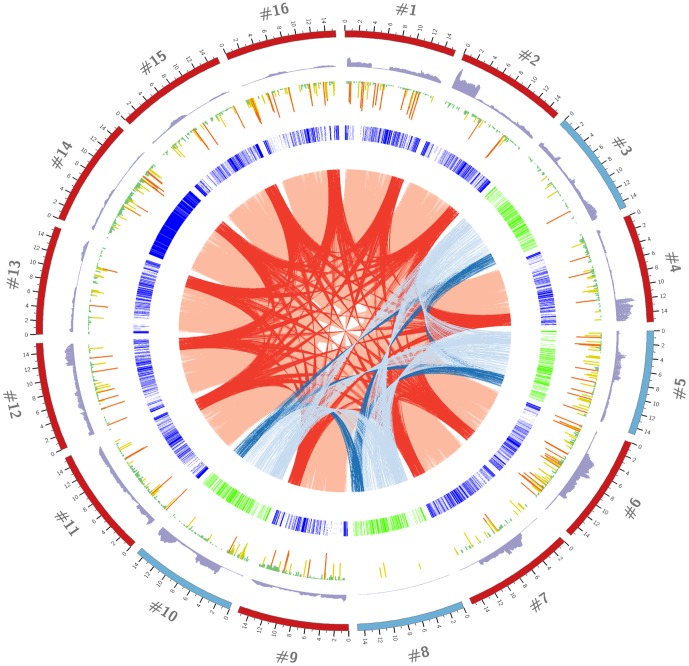
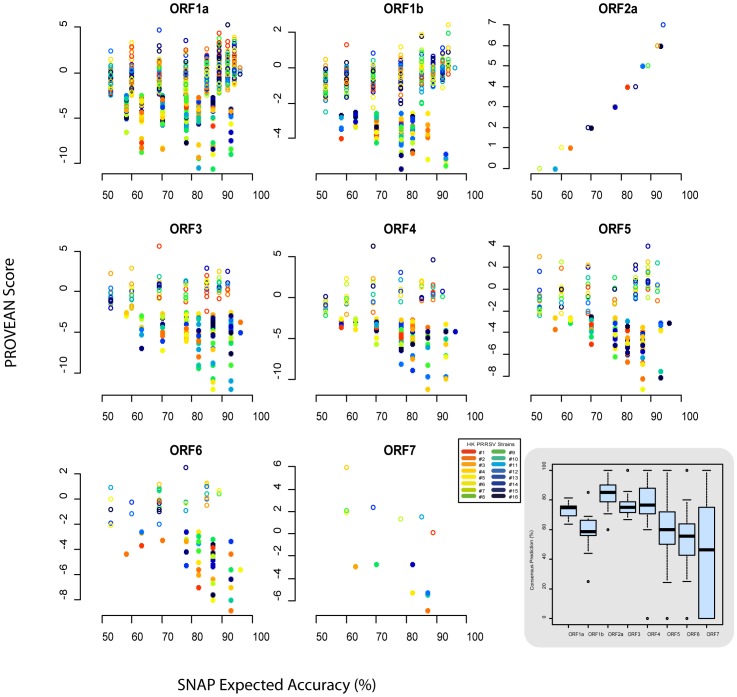
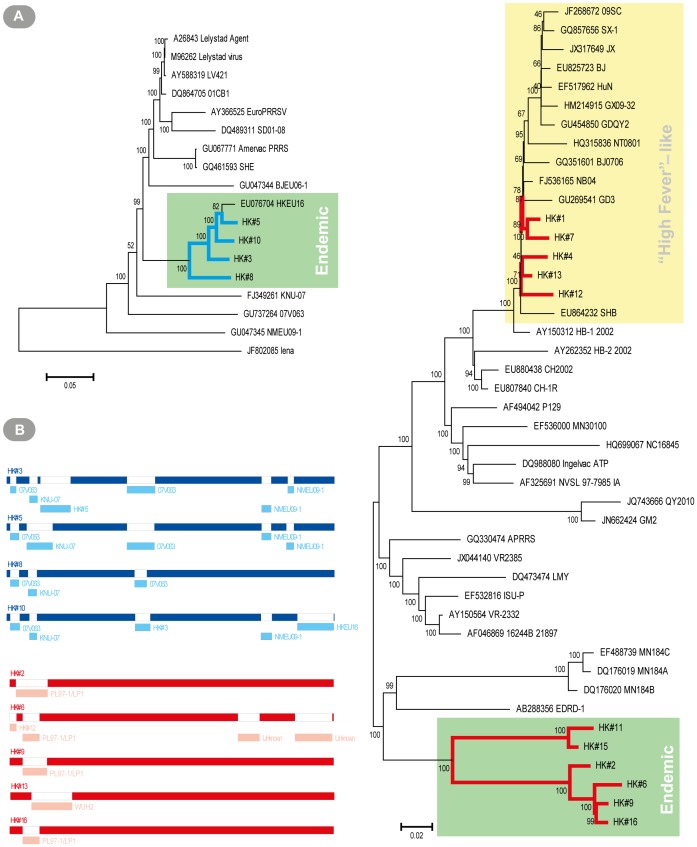
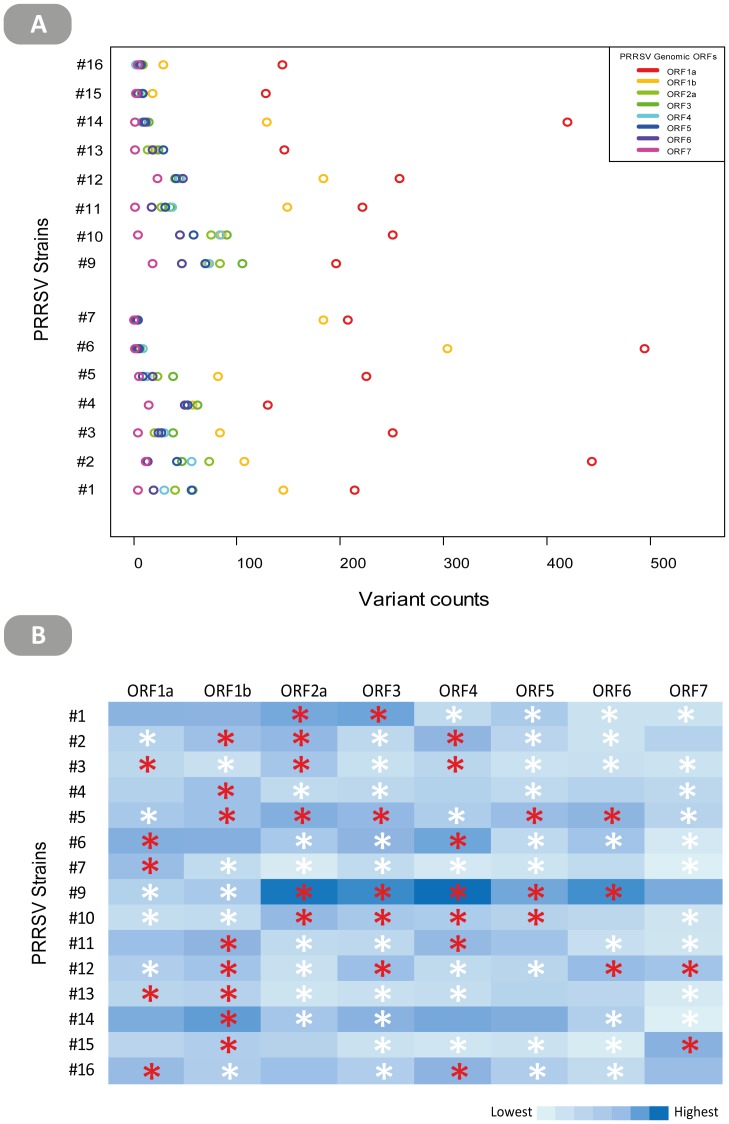
Most studies on PRRSV evolution have been limited to a particular region of the viral genome. A thorough genome-wide understanding of the impact of different mechanisms on shaping PRRSV genetic diversity is still lacking. To this end, deep sequencing was used to obtain genomic sequences of a diverse set of 16 isolates from a region of Hong Kong with a complex PRRSV epidemiological record. Genome assemblies and phylogenetic typing indicated the co-circulation of strains of both genotypes (type 1 and type 2) with varying Nsp2 deletion patterns and distinct evolutionary lineages ("High Fever"-like and local endemic type). Recombination analyses revealed genomic breakpoints in structural and non-structural regions of genomes of both genotypes with evidence of many recombination events originating from common ancestors. Additionally, the high fold of coverage per nucleotide allowed the characterization of minor variants arising from the quasispecies of each strain. Overall, 0.56-2.83% of sites were found to be polymorphic with respect to cognate consensus genomes. The distribution of minor variants across each genome was not uniform indicating the influence of selective forces. Proportion of variants capable of causing an amino acid change in their respective codons ranged between 25-67% with many predicted to be non-deleterious. Low frequency deletion variants were also detected providing one possible mechanism for their sudden emergence as cited in previous reports.
大多数关于猪繁殖与呼吸综合征病毒(PRRSV)进化的研究都局限于病毒基因组的特定区域。目前仍缺乏对不同机制在塑造PRRSV遗传多样性方面所产生影响的全面全基因组理解。为此,利用深度测序技术从香港一个具有复杂PRRSV流行病学记录的地区获取了16株不同分离株的基因组序列。基因组组装和系统发育分型表明,两种基因型(1型和2型)的毒株共同流行,具有不同的Nsp2缺失模式和不同的进化谱系(“高热”样和本地流行型)。重组分析揭示了两种基因型基因组的结构和非结构区域中的基因组断点,有证据表明许多重组事件源自共同祖先。此外,每个核苷酸的高覆盖倍数使得能够对每个毒株准种产生的次要变异进行表征。总体而言,相对于同源共有基因组,发现0.56 - 2.83%的位点是多态性的。次要变异在每个基因组中的分布并不均匀,表明存在选择力的影响。能够在各自密码子中引起氨基酸变化的变异比例在25 - 67%之间,许多被预测为无害。还检测到低频缺失变异,这为它们如先前报道中所述的突然出现提供了一种可能的机制。