Kamali Maryam, Marek Paul E, Peery Ashley, Antonio-Nkondjio Christophe, Ndo Cyrille, Tu Zhijian, Simard Frederic, Sharakhov Igor V
Department of Entomology, Virginia Polytechnic Institute and State University, Blacksburg, Virginia, United States of America.
Malaria Research Laboratory, OCEAC, Yaounde, Cameroon.
PLoS One. 2014 Apr 4;9(4):e93580. doi: 10.1371/journal.pone.0093580. eCollection 2014.
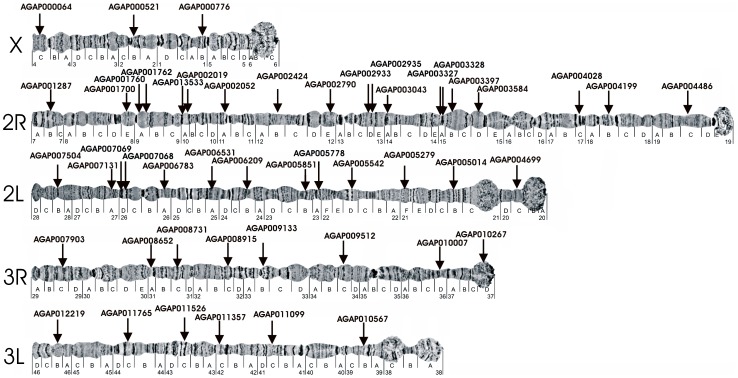
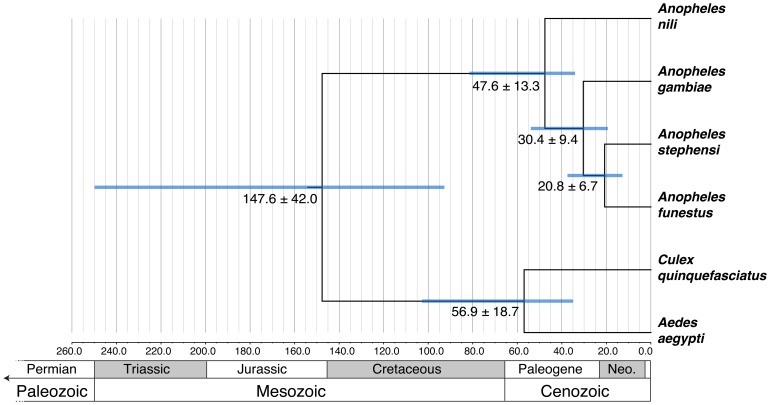
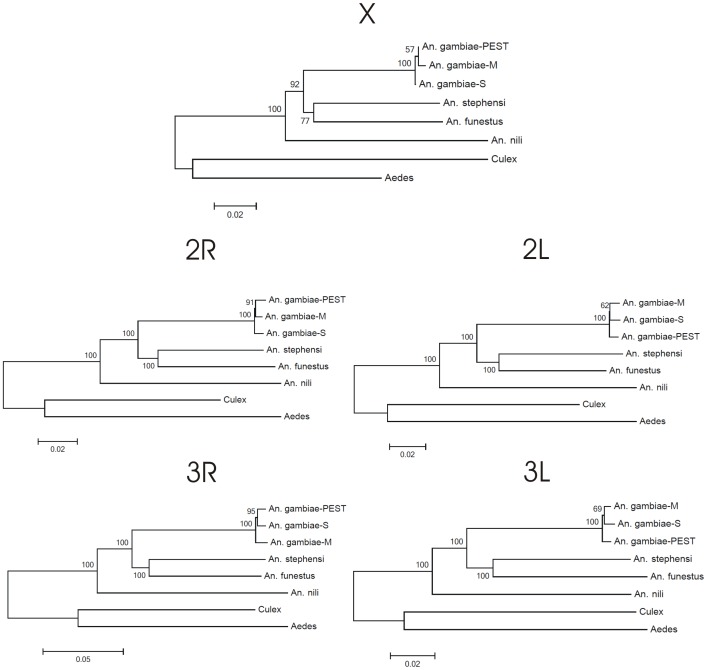
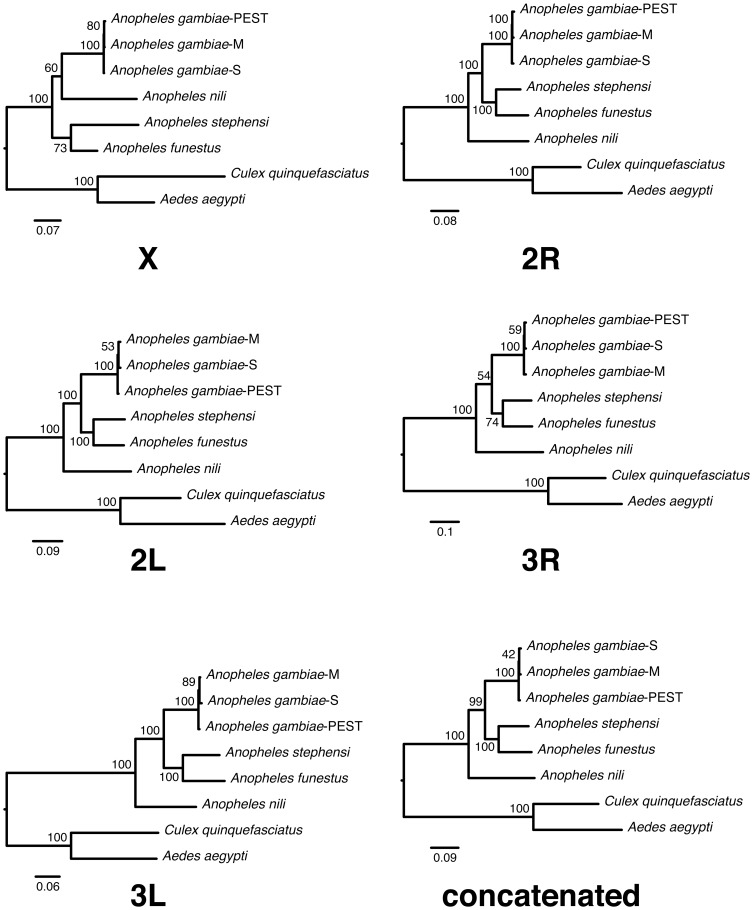
The major vectors of malaria in sub-Saharan Africa belong to subgenus Cellia. Yet, phylogenetic relationships and temporal diversification among African mosquito species have not been unambiguously determined. Knowledge about vector evolutionary history is crucial for correct interpretation of genetic changes identified through comparative genomics analyses. In this study, we estimated a molecular phylogeny using 49 gene sequences for the African malaria vectors An. gambiae, An. funestus, An. nili, the Asian malaria mosquito An. stephensi, and the outgroup species Culex quinquefasciatus and Aedes aegypti. To infer the phylogeny, we identified orthologous sequences uniformly distributed approximately every 5 Mb in the five chromosomal arms. The sequences were aligned and the phylogenetic trees were inferred using maximum likelihood and neighbor-joining methods. Bayesian molecular dating using a relaxed log normal model was used to infer divergence times. Trees from individual genes agreed with each other, placing An. nili as a basal clade that diversified from the studied malaria mosquito species 47.6 million years ago (mya). Other African malaria vectors originated more recently, and independently acquired traits related to vectorial capacity. The lineage leading to An. gambiae diverged 30.4 mya, while the African vector An. funestus and the Asian vector An. stephensi were the most closely related sister taxa that split 20.8 mya. These results were supported by consistently high bootstrap values in concatenated phylogenetic trees generated individually for each chromosomal arm. Genome-wide multigene phylogenetic analysis is a useful approach for discerning historic relationships among malaria vectors, providing a framework for the correct interpretation of genomic changes across species, and comprehending the evolutionary origins of this ubiquitous and deadly insect-borne disease.
撒哈拉以南非洲地区疟疾的主要传播媒介属于塞利疟蚊亚属。然而,非洲蚊种之间的系统发育关系和时间上的分化尚未得到明确确定。了解传播媒介的进化历史对于正确解释通过比较基因组学分析确定的基因变化至关重要。在本研究中,我们使用冈比亚按蚊、嗜人按蚊、尼罗按蚊这三种非洲疟疾传播媒介、亚洲疟疾蚊种斯氏按蚊以及外类群物种致倦库蚊和埃及伊蚊的49个基因序列估计了分子系统发育。为了推断系统发育,我们在五条染色体臂上确定了大致每隔5兆碱基均匀分布的直系同源序列。对这些序列进行比对,并使用最大似然法和邻接法推断系统发育树。使用宽松对数正态模型的贝叶斯分子定年法来推断分歧时间。来自各个基因的树相互一致,将尼罗按蚊置于一个基部进化枝中,该进化枝在4760万年前(mya)与所研究的疟疾蚊种分化。其他非洲疟疾传播媒介起源较晚,并独立获得了与传播能力相关的性状。导致冈比亚按蚊的谱系在3040万年前分化,而非洲传播媒介嗜人按蚊和亚洲传播媒介斯氏按蚊是关系最密切的姐妹分类群,在2080万年前分开。这些结果得到了为每个染色体臂单独生成的串联系统发育树中始终较高的自展值的支持。全基因组多基因系统发育分析是一种用于辨别疟疾传播媒介之间历史关系的有用方法,为正确解释物种间的基因组变化以及理解这种普遍存在且致命的虫媒疾病的进化起源提供了一个框架。