Kidd Jeffrey M, Sharpton Thomas J, Bobo Dean, Norman Paul J, Martin Alicia R, Carpenter Meredith L, Sikora Martin, Gignoux Christopher R, Nemat-Gorgani Neda, Adams Alexandra, Guadalupe Moraima, Guo Xiaosen, Feng Qiang, Li Yingrui, Liu Xiao, Parham Peter, Hoal Eileen G, Feldman Marcus W, Pollard Katherine S, Wall Jeffrey D, Bustamante Carlos D, Henn Brenna M
Department of Genetics, Stanford University, Stanford, CA 94305, USA.
BMC Genomics. 2014 Apr 4;15:262. doi: 10.1186/1471-2164-15-262.
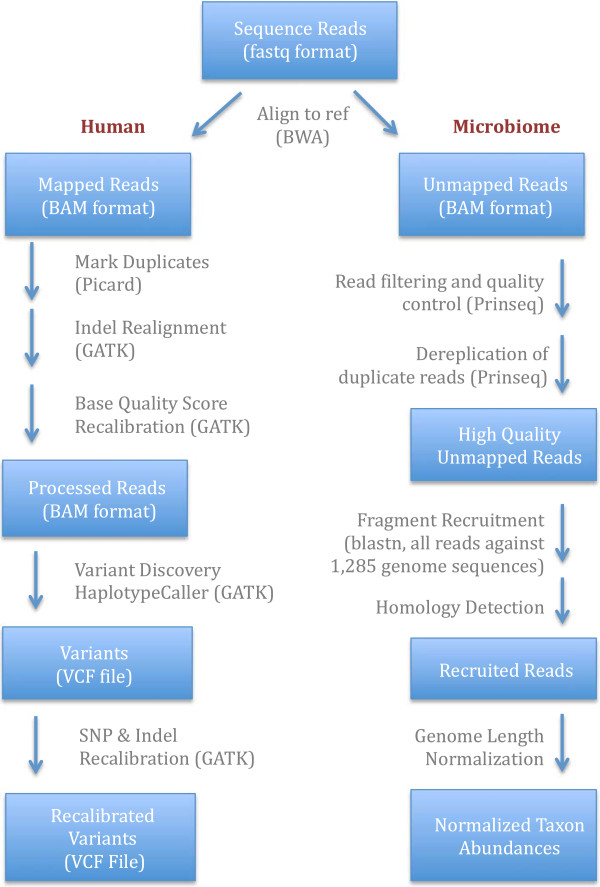
Targeted capture of genomic regions reduces sequencing cost while generating higher coverage by allowing biomedical researchers to focus on specific loci of interest, such as exons. Targeted capture also has the potential to facilitate the generation of genomic data from DNA collected via saliva or buccal cells. DNA samples derived from these cell types tend to have a lower human DNA yield, may be degraded from age and/or have contamination from bacteria or other ambient oral microbiota. However, thousands of samples have been previously collected from these cell types, and saliva collection has the advantage that it is a non-invasive and appropriate for a wide variety of research.
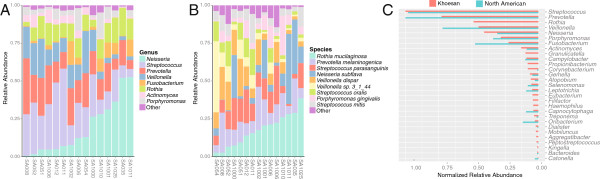
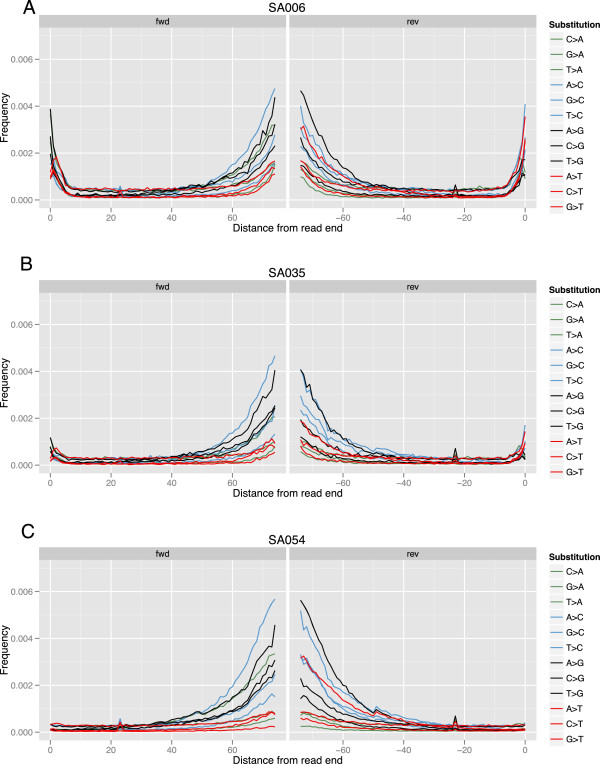
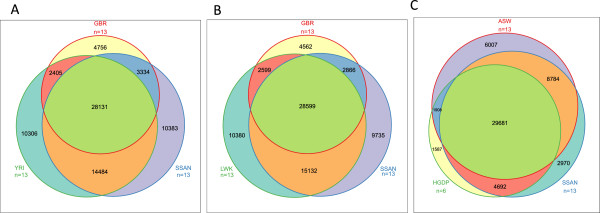
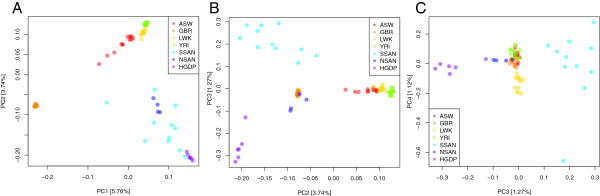
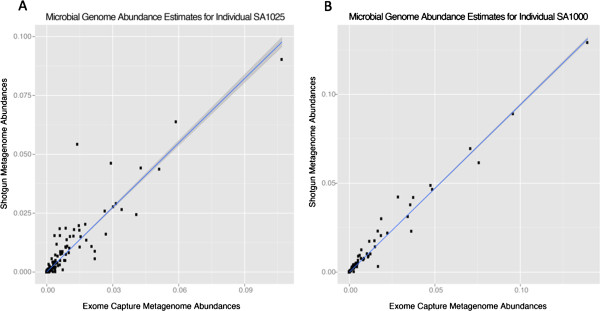
We demonstrate successful enrichment and sequencing of 15 South African KhoeSan exomes and 2 full genomes with samples initially derived from saliva. The expanded exome dataset enables us to characterize genetic diversity free from ascertainment bias for multiple KhoeSan populations, including new exome data from six HGDP Namibian San, revealing substantial population structure across the Kalahari Desert region. Additionally, we discover and independently verify thirty-one previously unknown KIR alleles using methods we developed to accurately map and call the highly polymorphic HLA and KIR loci from exome capture data. Finally, we show that exome capture of saliva-derived DNA yields sufficient non-human sequences to characterize oral microbial communities, including detection of bacteria linked to oral disease (e.g. Prevotella melaninogenica). For comparison, two samples were sequenced using standard full genome library preparation without exome capture and we found no systematic bias of metagenomic information between exome-captured and non-captured data.
DNA from human saliva samples, collected and extracted using standard procedures, can be used to successfully sequence high quality human exomes, and metagenomic data can be derived from non-human reads. We find that individuals from the Kalahari carry a higher oral pathogenic microbial load than samples surveyed in the Human Microbiome Project. Additionally, rare variants present in the exomes suggest strong population structure across different KhoeSan populations.
通过使生物医学研究人员能够专注于特定的感兴趣位点(如外显子),基因组区域的靶向捕获降低了测序成本,同时产生了更高的覆盖率。靶向捕获还有助于从通过唾液或颊细胞收集的DNA中生成基因组数据。源自这些细胞类型的DNA样本往往人类DNA产量较低,可能因时间而降解和/或受到细菌或其他口腔微生物群的污染。然而,此前已经从这些细胞类型中收集了数千个样本,并且唾液收集具有非侵入性且适用于广泛研究的优点。
我们展示了使用最初源自唾液的样本成功富集和测序15个南非科伊桑人的外显子组以及2个全基因组。扩展的外显子组数据集使我们能够在无确定偏差的情况下表征多个科伊桑人群的遗传多样性,包括来自六个HGDP纳米比亚桑人的新外显子组数据,揭示了喀拉哈里沙漠地区的大量种群结构。此外,我们使用我们开发的方法发现并独立验证了31个先前未知的杀伤细胞免疫球蛋白样受体(KIR)等位基因,该方法用于从外显子捕获数据中准确映射和调用高度多态的人类白细胞抗原(HLA)和KIR基因座。最后,我们表明源自唾液的DNA的外显子捕获产生了足够的非人类序列来表征口腔微生物群落,包括检测与口腔疾病相关的细菌(如产黑色素普雷沃菌)。为了进行比较,使用标准全基因组文库制备方法对两个样本进行了测序,未进行外显子捕获,并且我们发现外显子捕获数据和未捕获数据之间的宏基因组信息没有系统偏差。
使用标准程序收集和提取的人类唾液样本中的DNA可用于成功测序高质量的人类外显子组,并且宏基因组数据可从非人类读数中获得。我们发现喀拉哈里沙漠地区的个体比人类微生物组计划中调查的样本携带更高的口腔致病微生物负荷。此外,外显子组中存在的罕见变异表明不同科伊桑人群之间存在强大的种群结构。