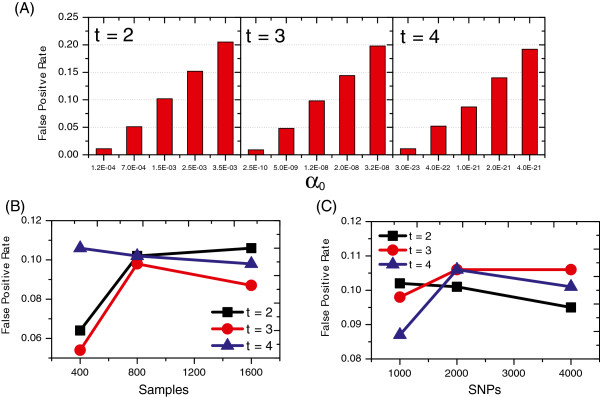
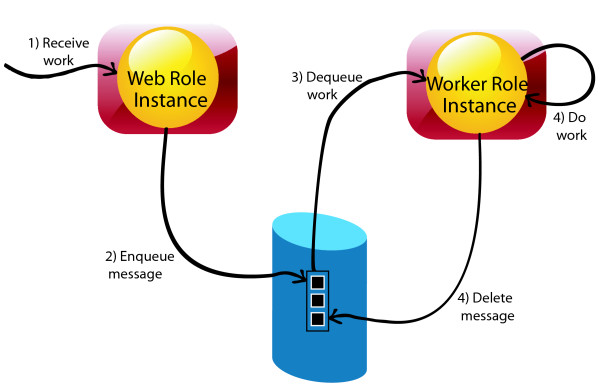
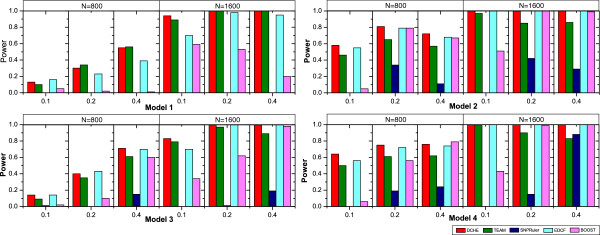
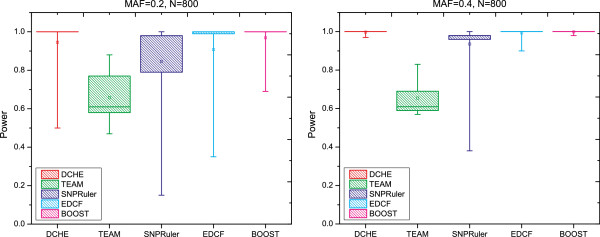
Department of Computer Science, Georgia State University, 34 Peachtree Street, Atlanta, USA.
BMC Bioinformatics. 2014 Apr 10;15:102. doi: 10.1186/1471-2105-15-102.
Taking the advantage of high-throughput single nucleotide polymorphism (SNP) genotyping technology, large genome-wide association studies (GWASs) have been considered to hold promise for unravelling complex relationships between genotype and phenotype. At present, traditional single-locus-based methods are insufficient to detect interactions consisting of multiple-locus, which are broadly existing in complex traits. In addition, statistic tests for high order epistatic interactions with more than 2 SNPs propose computational and analytical challenges because the computation increases exponentially as the cardinality of SNPs combinations gets larger.
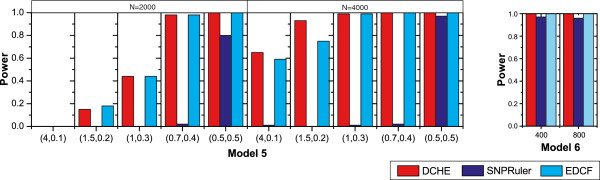
In this paper, we provide a simple, fast and powerful method using dynamic clustering and cloud computing to detect genome-wide multi-locus epistatic interactions. We have constructed systematic experiments to compare powers performance against some recently proposed algorithms, including TEAM, SNPRuler, EDCF and BOOST. Furthermore, we have applied our method on two real GWAS datasets, Age-related macular degeneration (AMD) and Rheumatoid arthritis (RA) datasets, where we find some novel potential disease-related genetic factors which are not shown up in detections of 2-loci epistatic interactions.
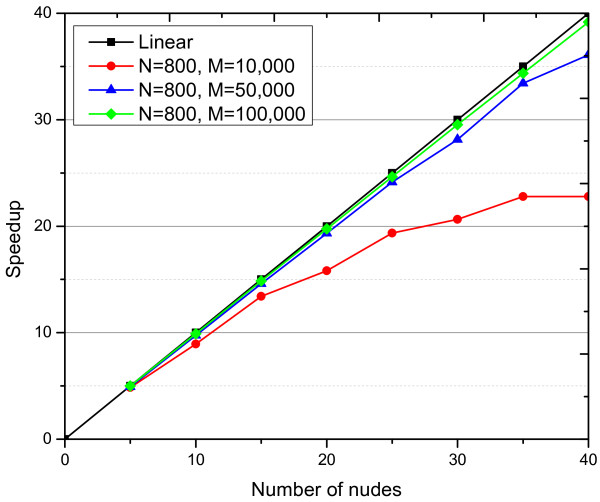
Experimental results on simulated data demonstrate that our method is more powerful than some recently proposed methods on both two- and three-locus disease models. Our method has discovered many novel high-order associations that are significantly enriched in cases from two real GWAS datasets. Moreover, the running time of the cloud implementation for our method on AMD dataset and RA dataset are roughly 2 hours and 50 hours on a cluster with forty small virtual machines for detecting two-locus interactions, respectively. Therefore, we believe that our method is suitable and effective for the full-scale analysis of multiple-locus epistatic interactions in GWAS.
利用高通量单核苷酸多态性(SNP)基因分型技术,全基因组关联研究(GWAS)被认为有望揭示基因型和表型之间的复杂关系。目前,传统的基于单基因座的方法不足以检测由多个基因座组成的相互作用,这些相互作用广泛存在于复杂特征中。此外,对于包含 2 个以上 SNP 的高阶上位性相互作用的统计检验提出了计算和分析方面的挑战,因为随着 SNP 组合的基数增大,计算量呈指数级增长。
在本文中,我们提供了一种简单、快速和强大的方法,使用动态聚类和云计算来检测全基因组多基因座上位性相互作用。我们已经构建了系统实验来比较与一些最近提出的算法的功效性能,包括 TEAM、SNPRuler、EDCF 和 BOOST。此外,我们将我们的方法应用于两个真实的 GWAS 数据集,即年龄相关性黄斑变性(AMD)和类风湿性关节炎(RA)数据集,在那里我们发现了一些新的潜在与疾病相关的遗传因素,这些因素在 2 个基因座上位性相互作用的检测中没有显示出来。
模拟数据的实验结果表明,我们的方法在两种和三种疾病模型上都比一些最近提出的方法更有效。我们的方法发现了许多新的高阶关联,这些关联在来自两个真实 GWAS 数据集的病例中显著富集。此外,我们的方法在 AMD 数据集和 RA 数据集上的云实现的运行时间大约分别为 2 小时和 50 小时,用于检测两个基因座的相互作用,在一个包含四十个小虚拟机的集群上。因此,我们相信我们的方法适用于全规模分析 GWAS 中的多基因座上位性相互作用。