Velasco Guillaume, Walton Emma L, Sterlin Delphine, Hédouin Sabrine, Nitta Hirohisa, Ito Yuya, Fouyssac Fanny, Mégarbané André, Sasaki Hiroyuki, Picard Capucine, Francastel Claire
Université Paris Diderot-Paris7, CNRS UMR7216, Epigénétique et Destin Cellulaire, Case Courrier 7042; 35, rue Hélène Brion, 75205 Paris, France.
Orphanet J Rare Dis. 2014 Apr 17;9:56. doi: 10.1186/1750-1172-9-56.
Immunodeficiency Centromeric Instability and Facial anomalies (ICF) is a rare autosomal recessive disease characterized by reduction in serum immunoglobulins with severe recurrent infections, facial dysmorphism, and more variable symptoms including mental retardation. ICF is directly related to a genomic methylation defect that mainly affects juxtacentromeric heterochromatin regions of certain chromosomes, leading to chromosomal rearrangements that constitute a hallmark of this syndrome upon cytogenetic testing. Mutations in the de novo DNA methyltransferase DNMT3B, the protein ZBTB24 of unknown function, or loci that remain to be identified, lie at its origin. Despite unifying features, common or distinguishing molecular signatures are still missing for this disease.
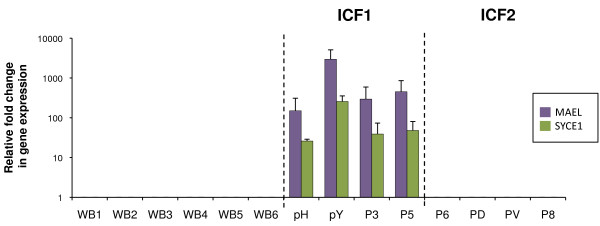
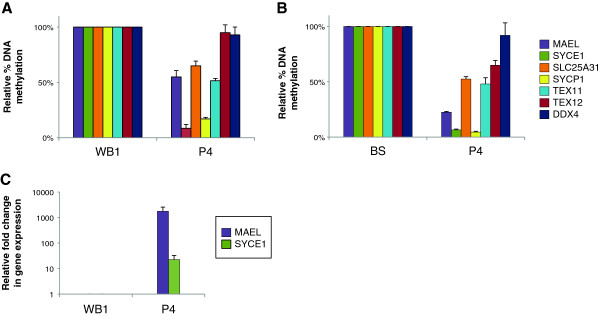
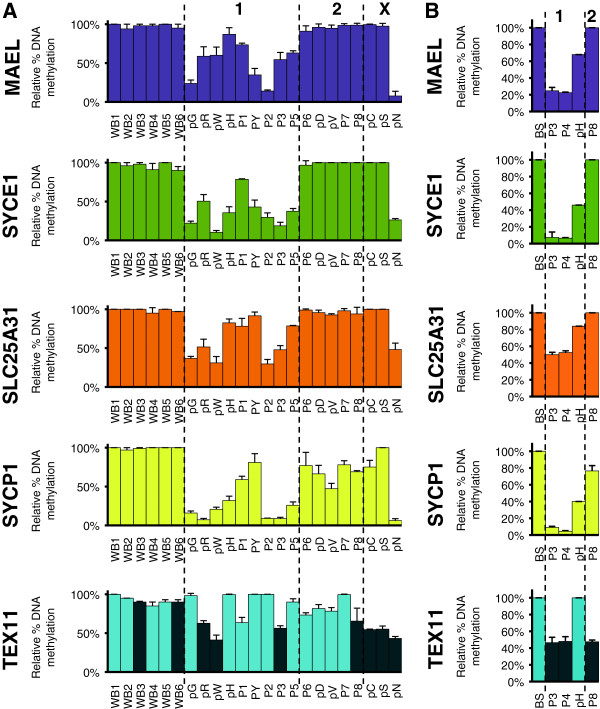
We used the molecular signature that we identified in a mouse model for ICF1 to establish transcriptional biomarkers to facilitate diagnosis and understanding of etiology of the disease. We assayed the expression and methylation status of a set of genes whose expression is normally restricted to germ cells, directly in whole blood samples and epithelial cells of ICF patients.
We report that DNA hypomethylation and expression of MAEL and SYCE1 represent robust biomarkers, easily testable directly from uncultured cells to diagnose the most prevalent sub-type of the syndrome. In addition, we identified the first unifying molecular signatures for ICF patients. Of importance, we validated the use of our biomarkers to diagnose a baby born to a family with a sick child. Finally, our analysis revealed unsuspected complex molecular signatures in two ICF patients suggestive of a novel genetic etiology for the disease.
Early diagnosis of ICF syndrome is crucial since early immunoglobulin supplementation can improve the course of disease. However, ICF is probably underdiagnosed, especially in patients that present with incomplete phenotype or born to families with no affected relatives. The specific and robust biomarkers identified in this study could be introduced into routine clinical immunology or neurology departments to facilitate testing of patients with suspected ICF syndrome. In addition, as exemplified by two patients with a combination of molecular defects never described before, our data support the search for new types of mutations at the origin of ICF syndrome.
免疫缺陷、着丝粒不稳定和面部异常(ICF)是一种罕见的常染色体隐性疾病,其特征为血清免疫球蛋白减少,伴有严重的反复感染、面部畸形,以及包括智力发育迟缓在内的更多可变症状。ICF与基因组甲基化缺陷直接相关,该缺陷主要影响某些染色体的近着丝粒异染色质区域,导致染色体重排,这在细胞遗传学检测中是该综合征的一个标志。从头DNA甲基转移酶DNMT3B、功能未知的蛋白质ZBTB24或有待确定的基因座中的突变是其病因。尽管具有统一特征,但该疾病仍缺乏共同或独特的分子特征。
我们使用在ICF1小鼠模型中鉴定出的分子特征来建立转录生物标志物,以促进疾病的诊断和病因理解。我们直接在ICF患者的全血样本和上皮细胞中检测了一组正常情况下仅在生殖细胞中表达的基因的表达和甲基化状态。
我们报告DNA低甲基化以及MAEL和SYCE1的表达代表了强大的生物标志物,可直接从未培养的细胞中轻松检测,以诊断该综合征最常见的亚型。此外,我们为ICF患者确定了首个统一的分子特征。重要的是,我们验证了使用我们的生物标志物诊断一个患病儿童家庭所生婴儿的情况。最后,我们的分析揭示了两名ICF患者中意想不到的复杂分子特征,提示该疾病存在新的遗传病因。
ICF综合征的早期诊断至关重要,因为早期补充免疫球蛋白可改善病程。然而,ICF可能未得到充分诊断,尤其是在表现出不完全表型的患者或出生于无患病亲属家庭的患者中。本研究中鉴定出的特异性和强大的生物标志物可引入常规临床免疫学或神经科,以方便对疑似ICF综合征的患者进行检测。此外,正如两名具有前所未有的分子缺陷组合的患者所示,我们的数据支持寻找ICF综合征起源的新型突变。