O'Connell Jared, Gurdasani Deepti, Delaneau Olivier, Pirastu Nicola, Ulivi Sheila, Cocca Massimiliano, Traglia Michela, Huang Jie, Huffman Jennifer E, Rudan Igor, McQuillan Ruth, Fraser Ross M, Campbell Harry, Polasek Ozren, Asiki Gershim, Ekoru Kenneth, Hayward Caroline, Wright Alan F, Vitart Veronique, Navarro Pau, Zagury Jean-Francois, Wilson James F, Toniolo Daniela, Gasparini Paolo, Soranzo Nicole, Sandhu Manjinder S, Marchini Jonathan
Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, United Kingdom; Department of Statistics, University of Oxford, Oxford, United Kingdom.
Wellcome Trust Sanger Institute, Hinxton, United Kingdom; Department of Public Health and Primary Care, University of Cambridge, Cambridge, United Kingdom.
PLoS Genet. 2014 Apr 17;10(4):e1004234. doi: 10.1371/journal.pgen.1004234. eCollection 2014 Apr.
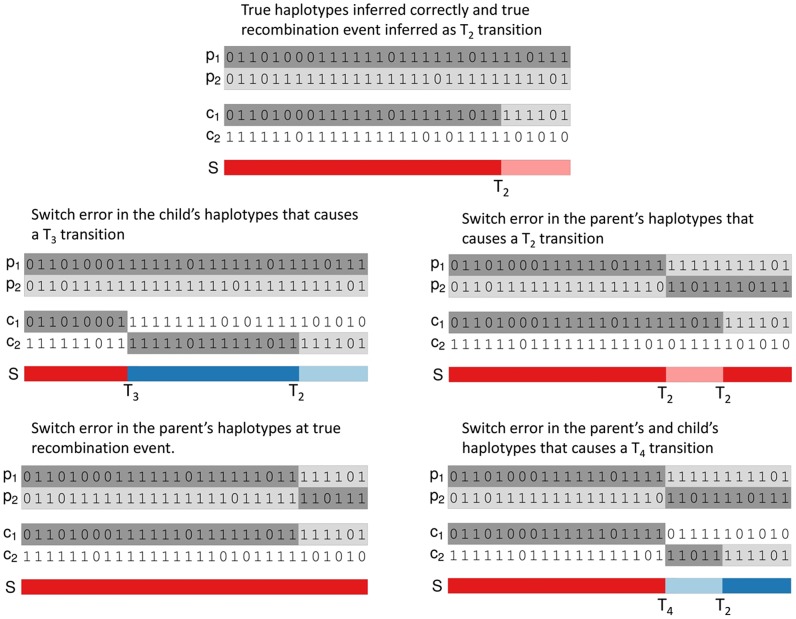
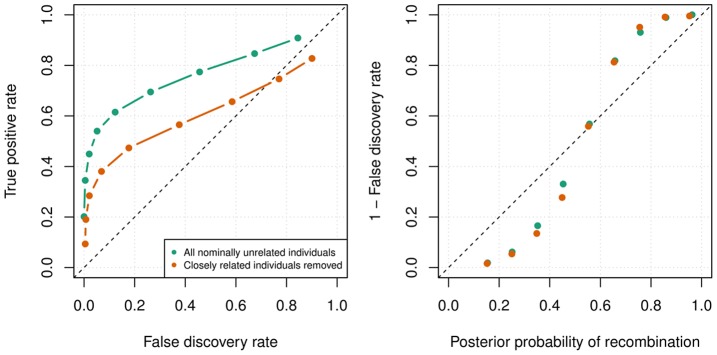
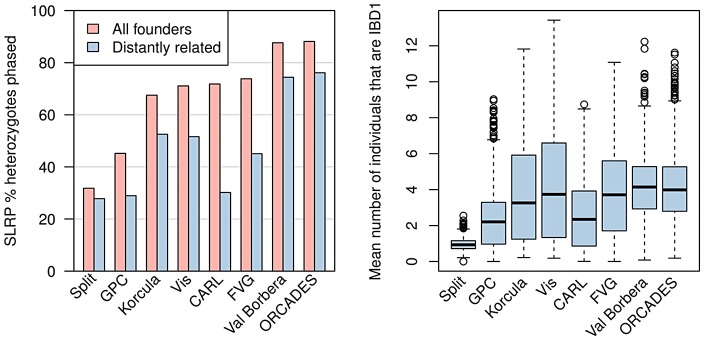
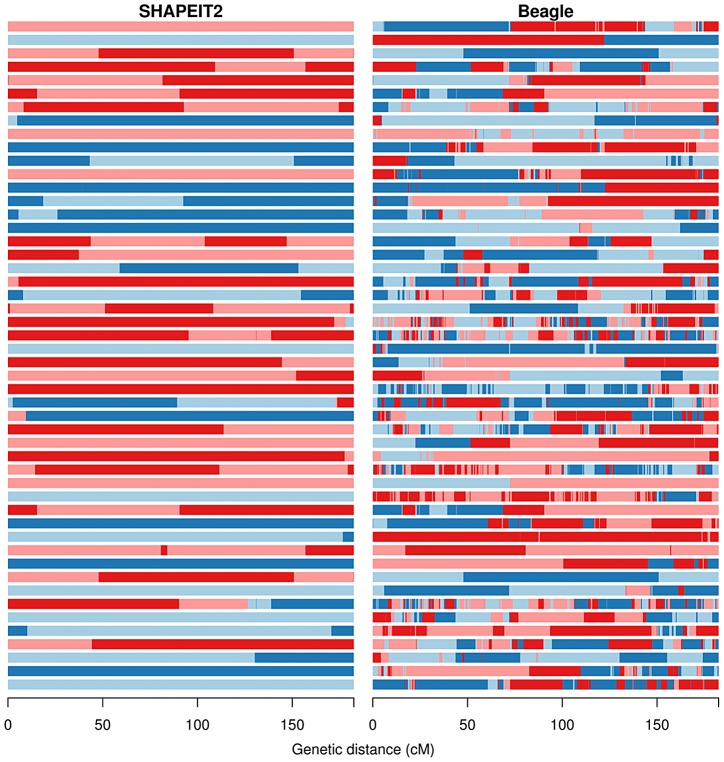
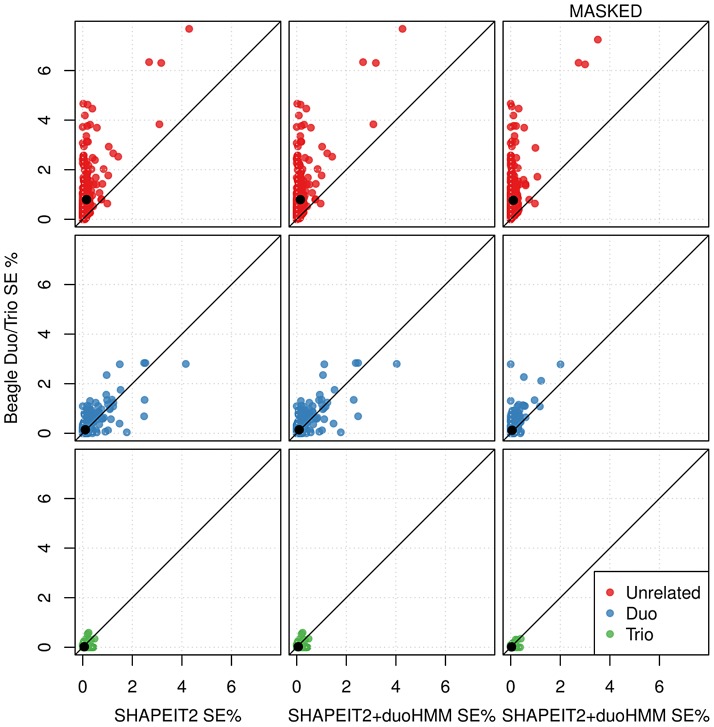
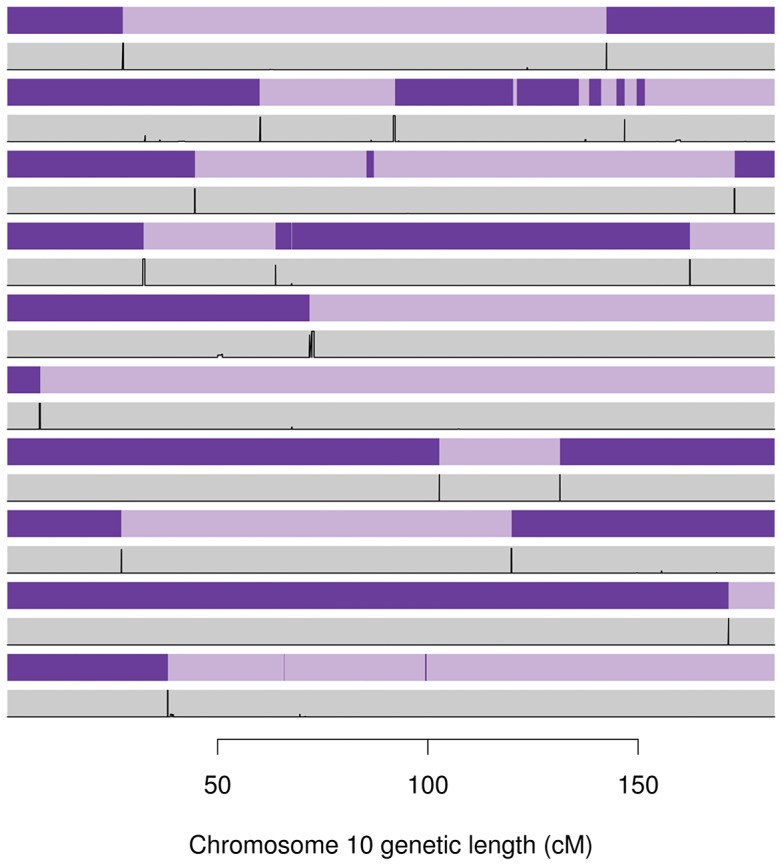
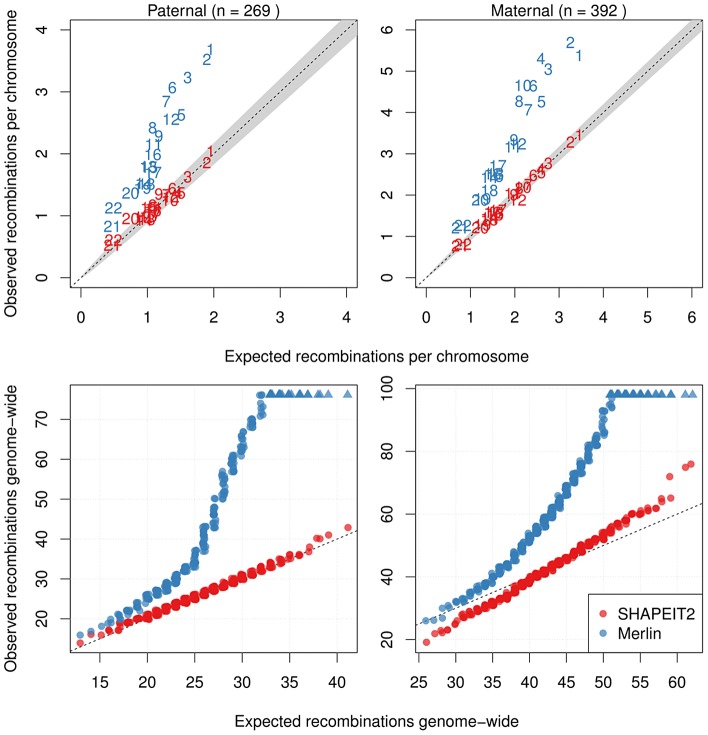
Many existing cohorts contain a range of relatedness between genotyped individuals, either by design or by chance. Haplotype estimation in such cohorts is a central step in many downstream analyses. Using genotypes from six cohorts from isolated populations and two cohorts from non-isolated populations, we have investigated the performance of different phasing methods designed for nominally 'unrelated' individuals. We find that SHAPEIT2 produces much lower switch error rates in all cohorts compared to other methods, including those designed specifically for isolated populations. In particular, when large amounts of IBD sharing is present, SHAPEIT2 infers close to perfect haplotypes. Based on these results we have developed a general strategy for phasing cohorts with any level of implicit or explicit relatedness between individuals. First SHAPEIT2 is run ignoring all explicit family information. We then apply a novel HMM method (duoHMM) to combine the SHAPEIT2 haplotypes with any family information to infer the inheritance pattern of each meiosis at all sites across each chromosome. This allows the correction of switch errors, detection of recombination events and genotyping errors. We show that the method detects numbers of recombination events that align very well with expectations based on genetic maps, and that it infers far fewer spurious recombination events than Merlin. The method can also detect genotyping errors and infer recombination events in otherwise uninformative families, such as trios and duos. The detected recombination events can be used in association scans for recombination phenotypes. The method provides a simple and unified approach to haplotype estimation, that will be of interest to researchers in the fields of human, animal and plant genetics.
许多现有的队列中,基因分型个体之间存在一定范围的亲缘关系,这或是出于设计,或是偶然因素。在此类队列中进行单倍型估计是许多下游分析的核心步骤。我们使用了来自隔离人群的六个队列以及来自非隔离人群的两个队列的基因型,研究了为名义上“无亲缘关系”个体设计的不同定相方法的性能。我们发现,与其他方法相比,包括专门为隔离人群设计的方法,SHAPEIT2在所有队列中产生的交换错误率要低得多。特别是当存在大量同源片段共享时,SHAPEIT2能推断出近乎完美的单倍型。基于这些结果,我们开发了一种通用策略,用于对个体间具有任何隐含或明确亲缘关系水平的队列进行定相。首先运行SHAPEIT2,忽略所有明确的家系信息。然后我们应用一种新颖的隐马尔可夫模型方法(duoHMM),将SHAPEIT2单倍型与任何家系信息相结合,以推断每条染色体上所有位点处每个减数分裂的遗传模式。这使得能够校正交换错误、检测重组事件和基因分型错误。我们表明,该方法检测到的重组事件数量与基于遗传图谱的预期非常吻合,并且它推断出的虚假重组事件比Merlin少得多。该方法还可以检测基因分型错误,并在其他信息不足的家系(如三联体和二元体)中推断重组事件。检测到的重组事件可用于重组表型的关联扫描。该方法为单倍型估计提供了一种简单统一的方法,将对人类、动物和植物遗传学领域的研究人员具有吸引力。