Angelis Konstantinos, Dos Reis Mario, Yang Ziheng
Department of Genetics, Evolution and Environment, University College London, London, United Kingdom.
Department of Genetics, Evolution and Environment, University College London, London, United Kingdom
Mol Biol Evol. 2014 Jul;31(7):1902-13. doi: 10.1093/molbev/msu142. Epub 2014 Apr 18.
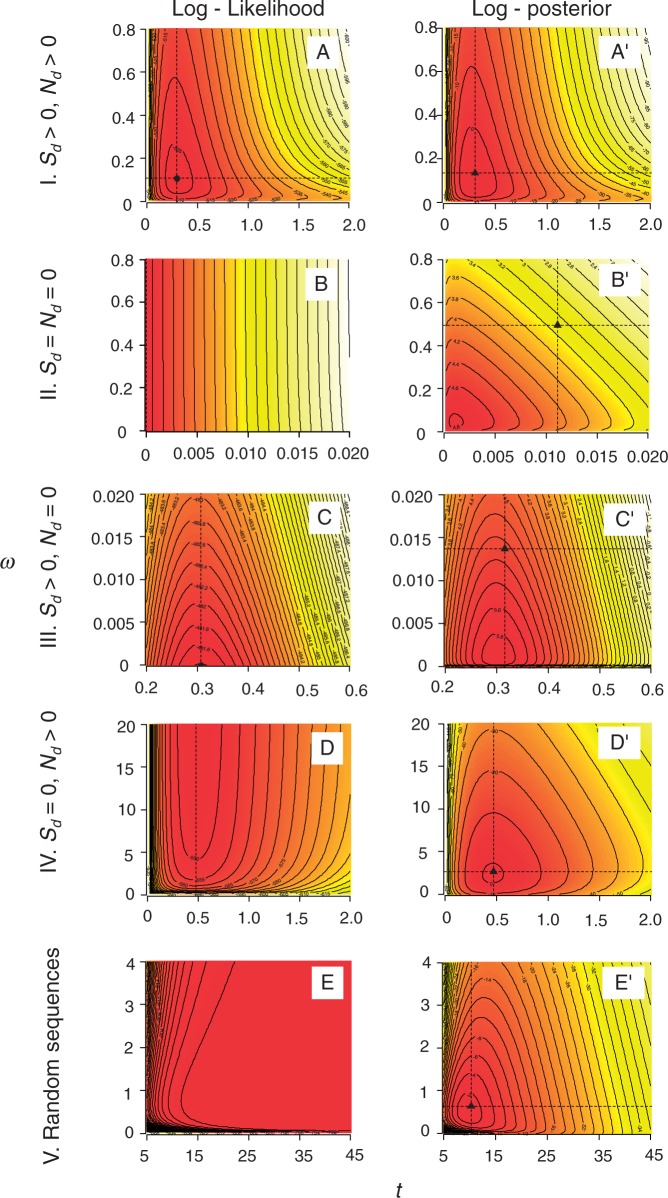
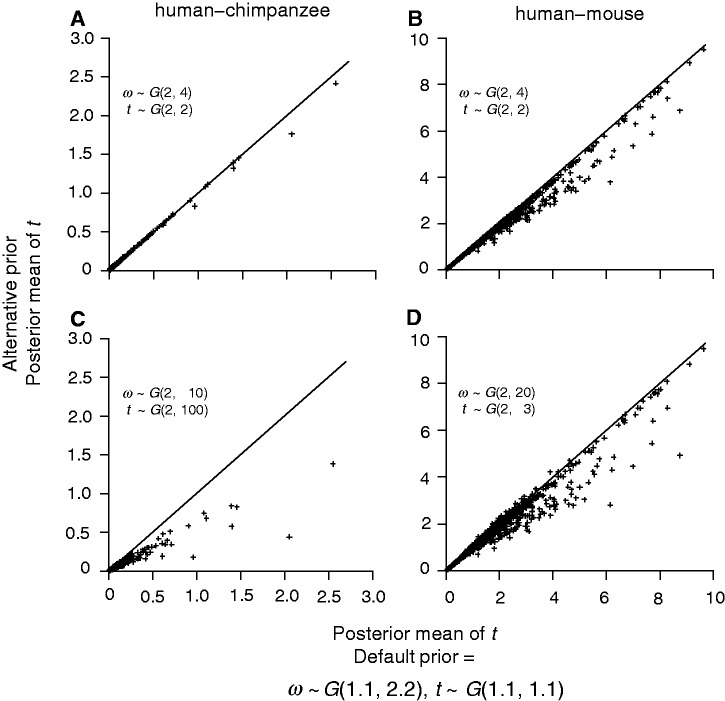
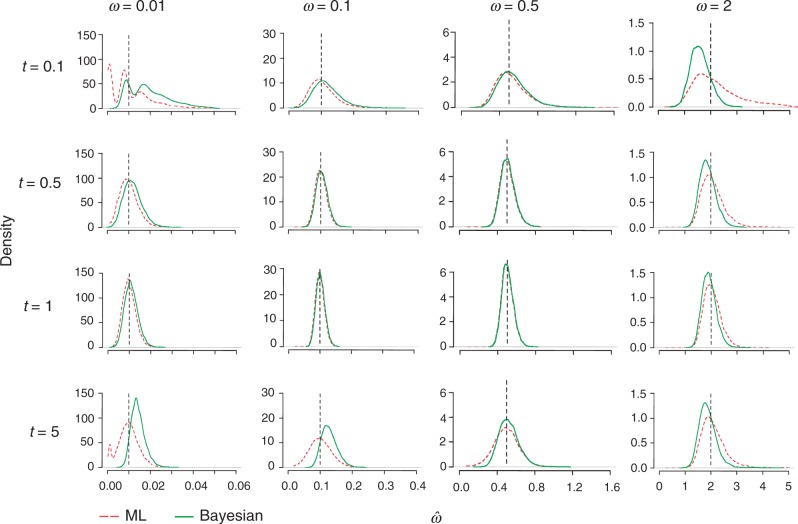
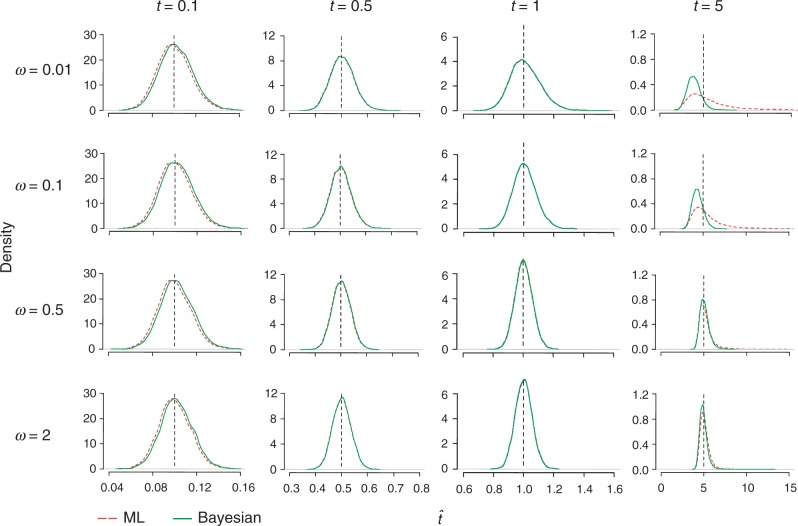
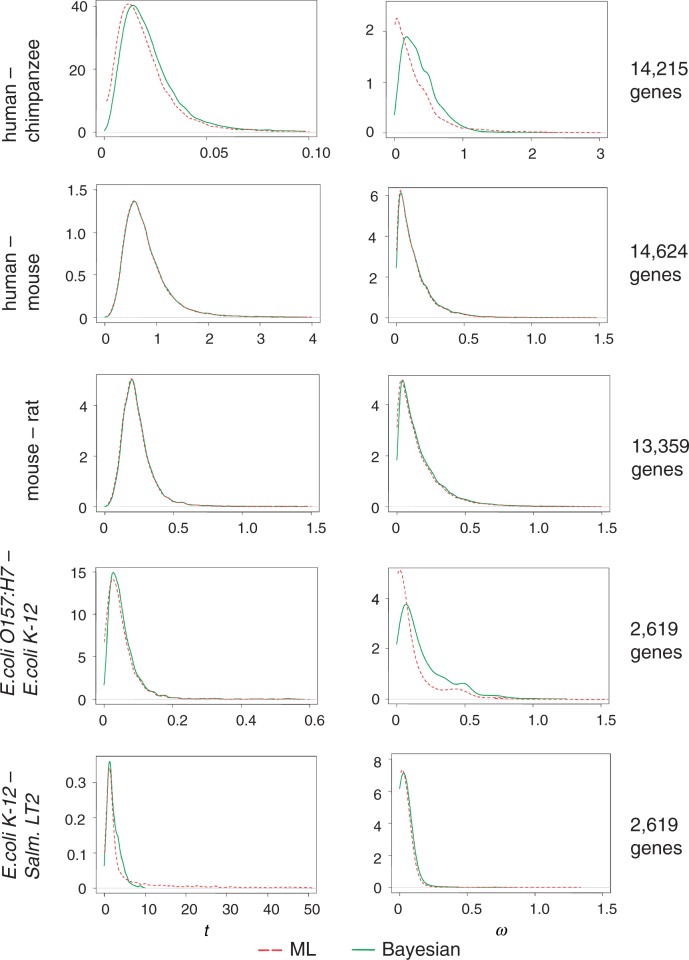
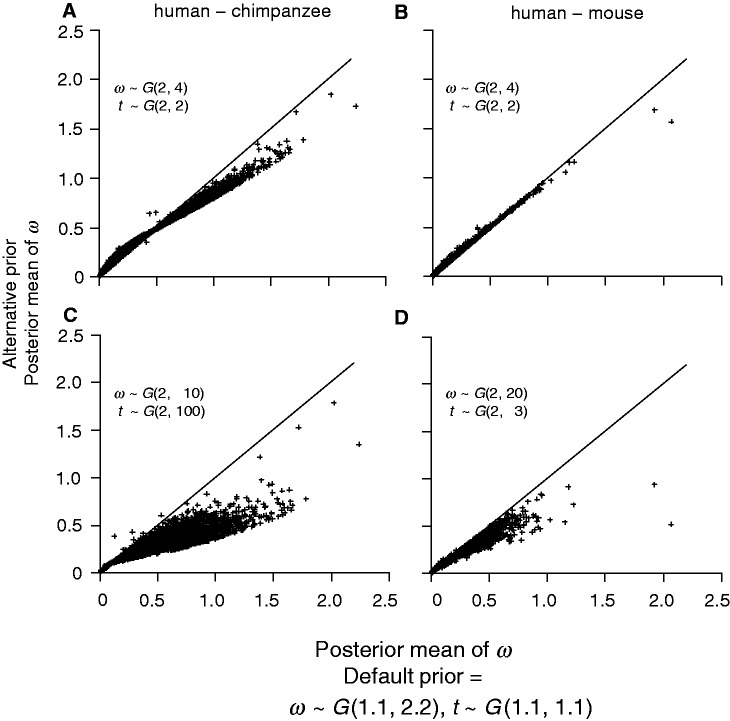
The nonsynonymous/synonymous rate ratio (ω = d(N)/d(S)) is an important measure of the mode and strength of natural selection acting on nonsynonymous mutations in protein-coding genes. The simplest such analysis is the estimation of the d(N)/d(S) ratio using two sequences. Both heuristic counting methods and the maximum-likelihood (ML) method based on a codon substitution model are widely used for such analysis. However, these methods do not have nice statistical properties, as the estimates can be zero or infinity in some data sets, so that their means and variances are infinite. In large genome-scale comparisons, such extreme estimates (either 0 or ∞) of ω and sequence distance (t) are common. Here, we implement a Bayesian method to estimate ω and t in pairwise sequence comparisons. Using a combination of computer simulation and real data analysis, we show that the Bayesian estimates have better statistical properties than the ML estimates, because the prior on ω and t shrinks the posterior of those parameters away from extreme values. We also calculate the posterior probability for ω > 1 as a Bayesian alternative to the likelihood ratio test. The new method is computationally efficient and may be useful for genome-scale comparisons of protein-coding gene sequences.
非同义/同义比率(ω = d(N)/d(S))是衡量作用于蛋白质编码基因中非同义突变的自然选择模式和强度的重要指标。最简单的此类分析是使用两个序列估计d(N)/d(S)比率。启发式计数方法和基于密码子替换模型的最大似然(ML)方法都广泛用于此类分析。然而,这些方法没有良好的统计特性,因为在某些数据集中估计值可能为零或无穷大,因此它们的均值和方差是无穷的。在大规模基因组比较中,ω和序列距离(t)的此类极端估计值(0或∞)很常见。在这里,我们实现了一种贝叶斯方法来估计成对序列比较中的ω和t。通过计算机模拟和实际数据分析相结合,我们表明贝叶斯估计比ML估计具有更好的统计特性,因为ω和t的先验将这些参数的后验从极端值收缩回来。我们还计算了ω > 1的后验概率,作为似然比检验的贝叶斯替代方法。新方法计算效率高,可能对蛋白质编码基因序列的基因组规模比较有用。