University of Liège and Centre Hospitalier Universitaire (CHU) Sart-Tilman, Liège, Belgium.
Arthritis Rheumatol. 2014 Apr;66(4):960-8. doi: 10.1002/art.38315.
To compare the gene expression patterns of synovial cells from inflamed or normal/reactive areas of synovial membrane obtained from the same patient with osteoarthritis (OA).
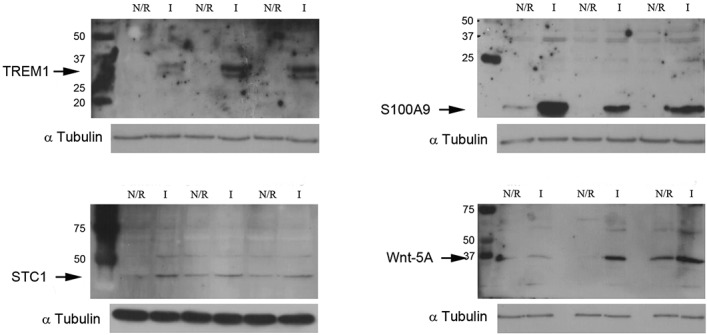
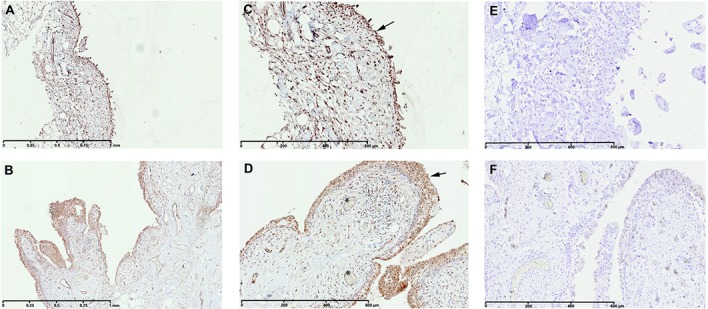
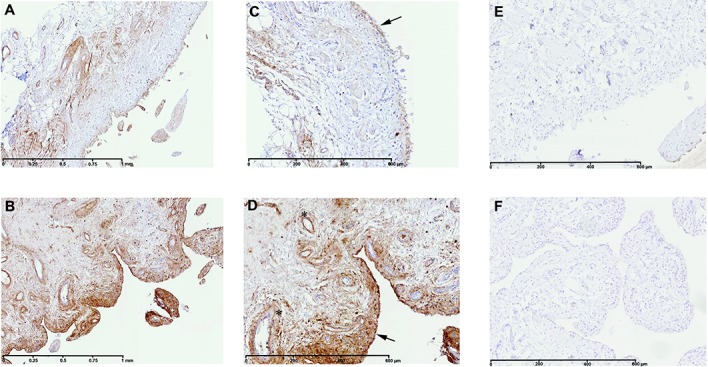
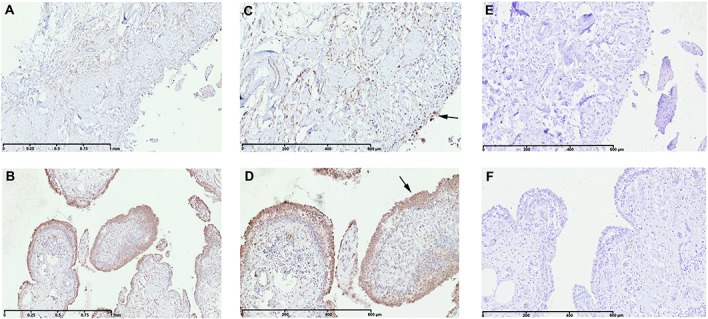
At the time of total knee replacement, synovial tissues were obtained from 12 patients with knee OA. The inflammation status of the synovial membrane was characterized according to macroscopic criteria and classified as normal/reactive or inflamed. Biopsy samples were cultured separately for 7 days. Microarray gene expression profiling was performed on normal/reactive and inflamed areas. Western blot and immunohistochemistry were used to confirm the identified genes that were differentially expressed.
We identified 896 genes that were differentially expressed between normal/reactive and inflamed areas. The key pathways were related to inflammation, cartilage metabolism, Wnt signaling, and angiogenesis. In the inflammation network, the genes TREM1 and S100A9 were strongly up-regulated. The genes MMP3, MMP9, CTSH (cathepsin H), and CTSS (cathepsin S) were significantly up-regulated in the cartilage catabolism pathway, while the most up-regulated anabolism enzyme gene was HAS1. In the Wnt signaling pathway, the genes for Wnt-5a and low-density lipoprotein receptor-related protein 5 were up-regulated, while the gene FZD2 and the gene for Dkk-3 were down-regulated. Finally, STC1, which codes for a protein involved in angiogenesis, was identified as the most up-regulated gene in inflamed compared with normal/reactive areas.
This study is the first to identify different expression patterns between 2 areas of the synovial membrane from the same patient. These differences concern several key pathways involved in OA pathogenesis. This analysis also provides information regarding new genes and proteins as potential targets of treatment.
比较来自同一膝骨关节炎(OA)患者的滑膜炎症或正常/反应区滑膜细胞的基因表达模式。
在全膝关节置换时,从 12 例膝 OA 患者的滑膜组织中获取滑膜组织。根据宏观标准对滑膜膜的炎症状态进行特征描述,并分为正常/反应性或炎症性。将活检样本分别培养 7 天。对正常/反应性和炎症性区域进行微阵列基因表达谱分析。使用 Western blot 和免疫组织化学法来验证差异表达的鉴定基因。
我们鉴定了 896 个在正常/反应性和炎症性区域之间差异表达的基因。关键途径与炎症、软骨代谢、Wnt 信号传导和血管生成有关。在炎症网络中,TREM1 和 S100A9 基因强烈上调。在软骨代谢途径中,MMP3、MMP9、CTSH(组织蛋白酶 H)和 CTSS(组织蛋白酶 S)基因显著上调,而上调最明显的代谢酶基因是 HAS1。在 Wnt 信号通路中,Wnt-5a 和低密度脂蛋白受体相关蛋白 5 的基因上调,而 FZD2 基因和 Dkk-3 基因下调。最后,编码参与血管生成的蛋白的 STC1 基因被鉴定为与正常/反应区相比在炎症区中上调最明显的基因。
本研究首次鉴定了来自同一患者的滑膜膜 2 个区域之间的不同表达模式。这些差异涉及到 OA 发病机制中几个关键途径。该分析还提供了有关新基因和蛋白作为潜在治疗靶点的信息。