Deak Andras T, Blass Sandra, Khan Muhammad J, Groschner Lukas N, Waldeck-Weiermair Markus, Hallström Seth, Graier Wolfgang F, Malli Roland
The Institute of Molecular Biology and Biochemistry, Center of Molecular Medicine, Medical University of Graz, 8010-Graz, Austria.
The Institute of Physiological Chemistry, Center of Physiological Medicine, Medical University of Graz, 8010-Graz, Austria.
J Cell Sci. 2014 Jul 1;127(Pt 13):2944-55. doi: 10.1242/jcs.149807. Epub 2014 May 7.
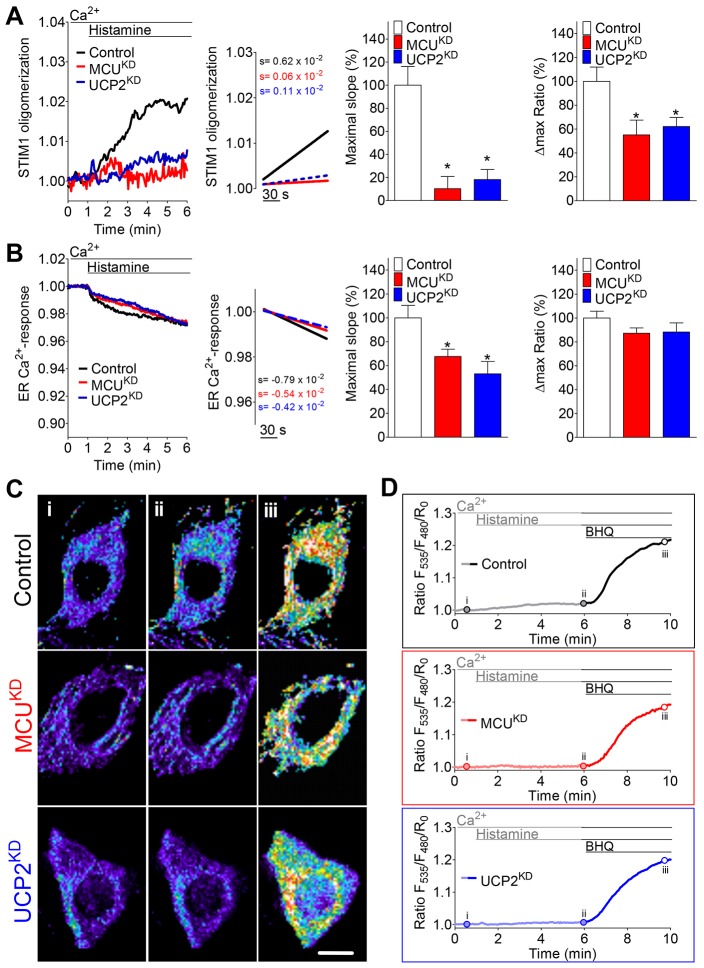
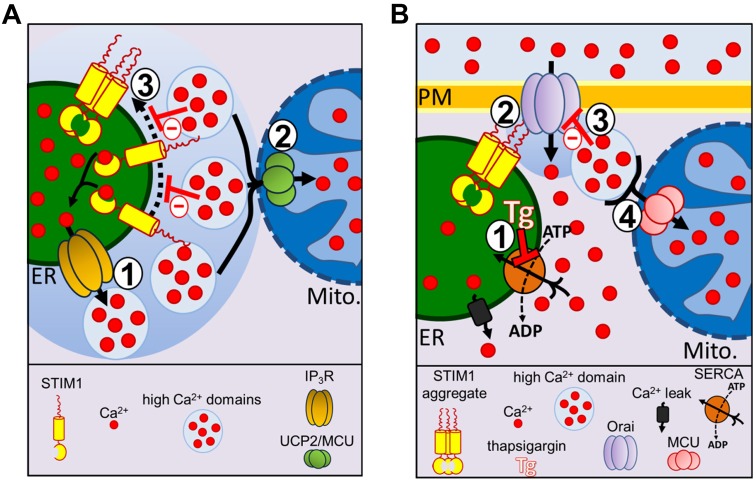
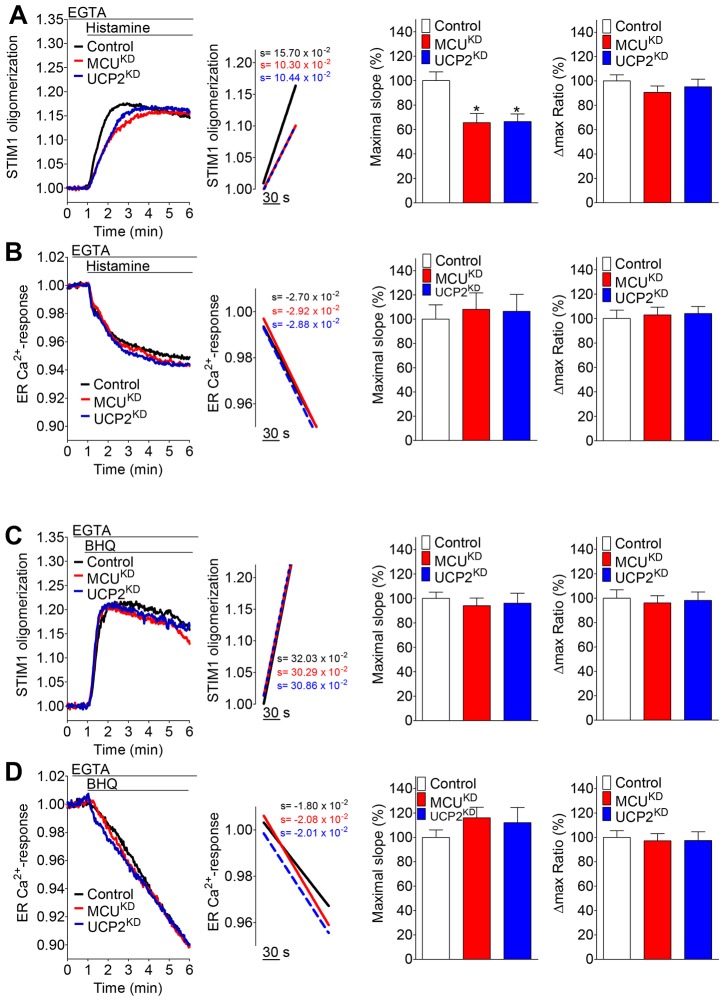
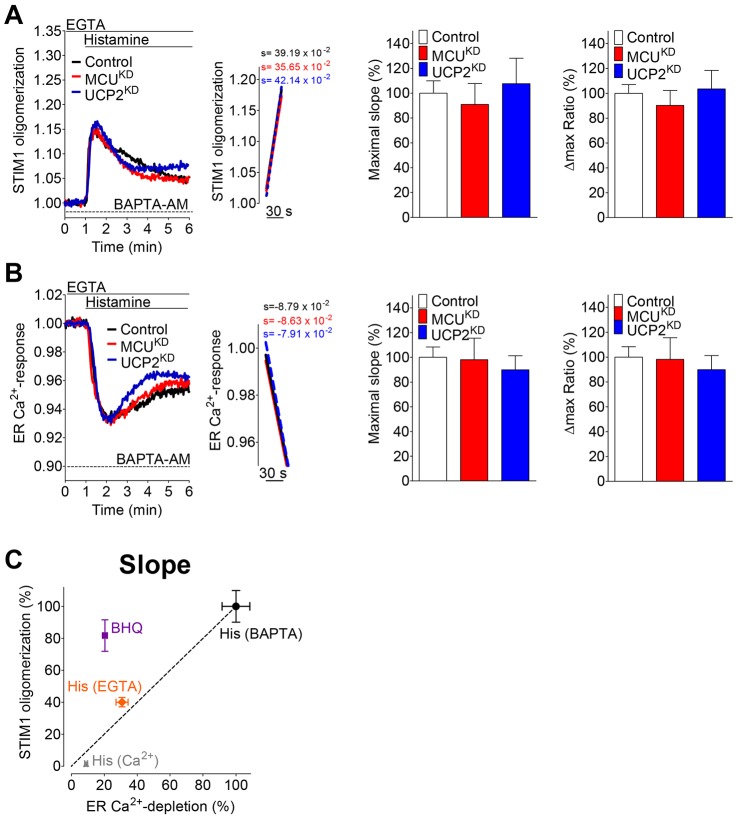
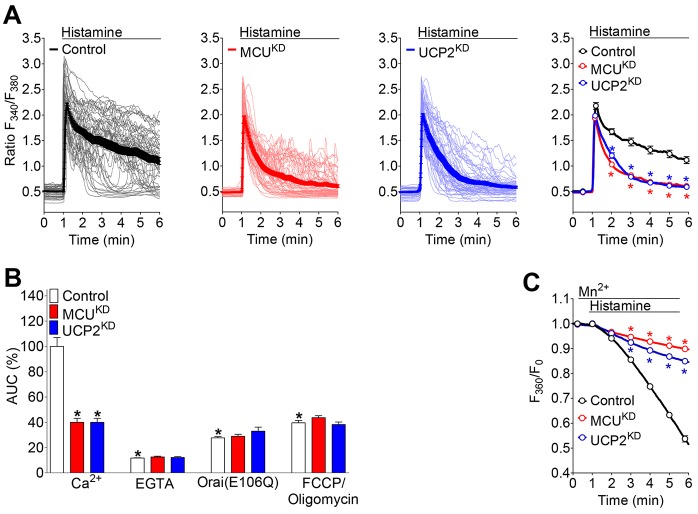
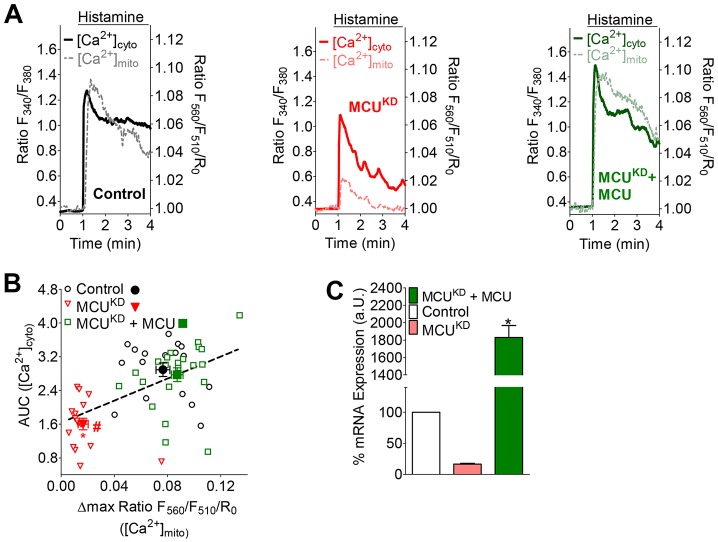
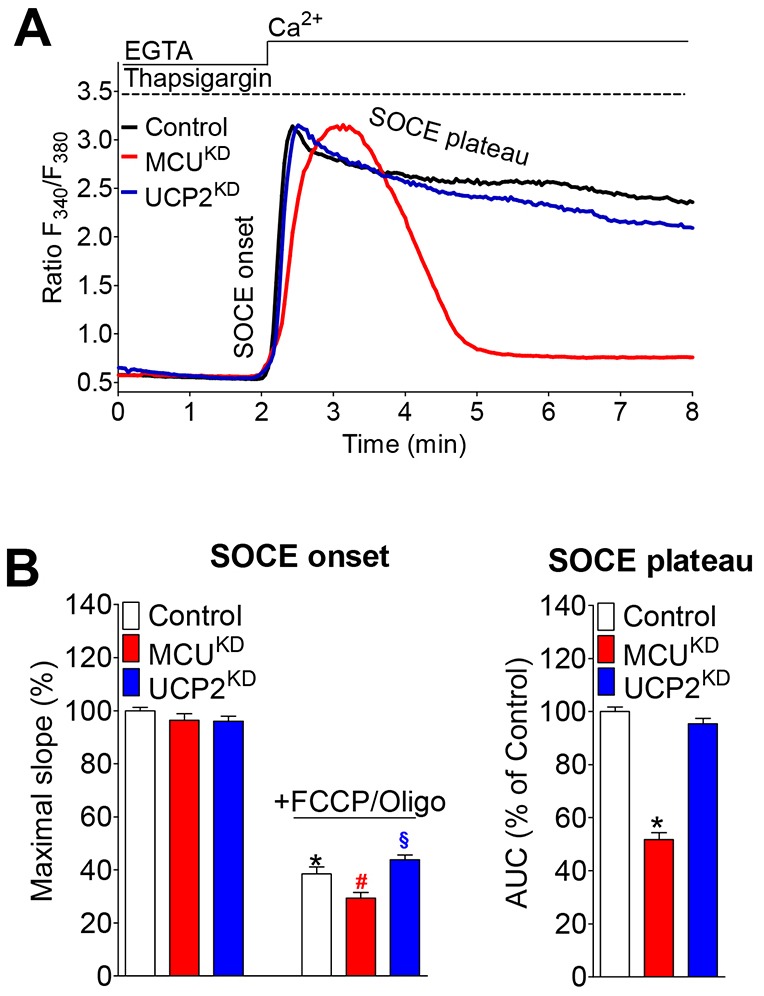
Mitochondria contribute to cell signaling by controlling store-operated Ca(2+) entry (SOCE). SOCE is activated by Ca(2+) release from the endoplasmic reticulum (ER), whereupon stromal interacting molecule 1 (STIM1) forms oligomers, redistributes to ER-plasma-membrane junctions and opens plasma membrane Ca(2+) channels. The mechanisms by which mitochondria interfere with the complex process of SOCE are insufficiently clarified. In this study, we used an shRNA approach to investigate the direct involvement of mitochondrial Ca(2+) buffering in SOCE. We demonstrate that knockdown of either of two proteins that are essential for mitochondrial Ca(2+) uptake, the mitochondrial calcium uniporter (MCU) or uncoupling protein 2 (UCP2), results in decelerated STIM1 oligomerization and impaired SOCE following cell stimulation with an inositol-1,4,5-trisphosphate (IP3)-generating agonist. Upon artificially augmented cytosolic Ca(2+) buffering or ER Ca(2+) depletion by sarcoplasmic or endoplasmic reticulum Ca(2+)-ATPase (SERCA) inhibitors, STIM1 oligomerization did not rely on intact mitochondrial Ca(2+) uptake. However, MCU-dependent mitochondrial sequestration of Ca(2+) entering through the SOCE pathway was essential to prevent slow deactivation of SOCE. Our findings show a stimulus-specific contribution of mitochondrial Ca(2+) uptake to the SOCE machinery, likely through a role in shaping cytosolic Ca(2+) micro-domains.
线粒体通过控制储存性钙(Ca2+)内流(SOCE)参与细胞信号传导。SOCE由内质网(ER)释放Ca2+激活,随后基质相互作用分子1(STIM1)形成寡聚体,重新分布到内质网 - 质膜连接处并打开质膜Ca2+通道。线粒体干扰SOCE复杂过程的机制尚未完全阐明。在本研究中,我们使用短发夹RNA(shRNA)方法来研究线粒体Ca2+缓冲在SOCE中的直接作用。我们证明,敲低线粒体Ca2+摄取所必需的两种蛋白质中的任何一种,即线粒体钙单向转运体(MCU)或解偶联蛋白2(UCP2),在用生成肌醇 - 1,4,5 - 三磷酸(IP3)的激动剂刺激细胞后,会导致STIM1寡聚化减速和SOCE受损。在用肌浆网或内质网Ca2+ - ATP酶(SERCA)抑制剂人为增加胞质Ca2+缓冲或内质网Ca2+耗竭后,STIM1寡聚化不依赖于完整的线粒体Ca2+摄取。然而,MCU依赖的通过SOCE途径进入的Ca2+的线粒体隔离对于防止SOCE的缓慢失活至关重要。我们的研究结果表明,线粒体Ca2+摄取对SOCE机制具有刺激特异性作用,可能是通过在塑造胞质Ca2+微区中的作用来实现的。