Department of Medicine and Program for Personalized and Genomic Medicine, University of Maryland School of Medicine Baltimore, MD, USA ; Veterans Administration Medical Center Baltimore, MD, USA.
Department of Medicine, University of Texas Health Science Center Houston, TX, USA.
Front Genet. 2014 Apr 29;5:95. doi: 10.3389/fgene.2014.00095. eCollection 2014.
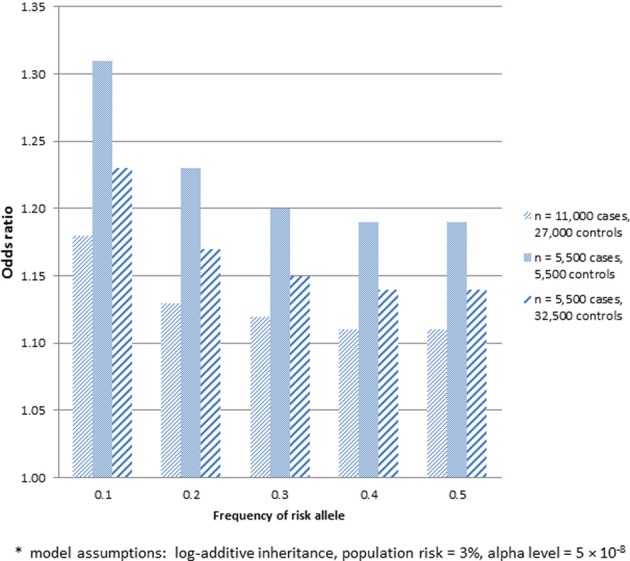
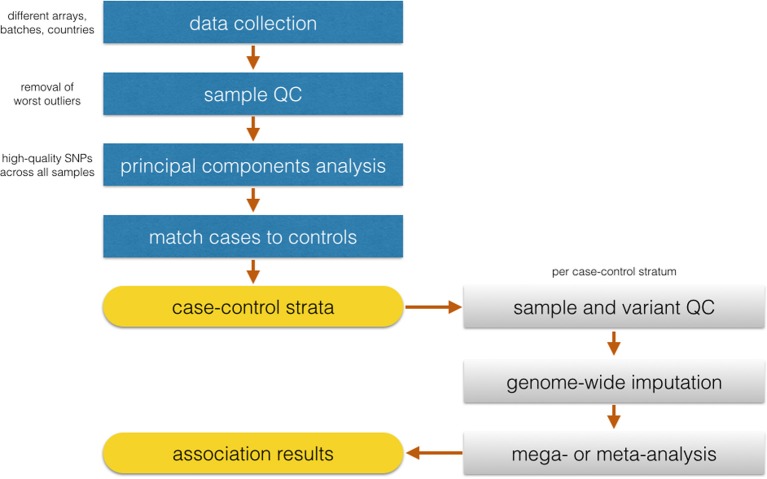
Genome-wide association studies (GWAS) are widely applied to identify susceptibility loci for a variety of diseases using genotyping arrays that interrogate known polymorphisms throughout the genome. A particular strength of GWAS is that it is unbiased with respect to specific genomic elements (e.g., coding or regulatory regions of genes), and it has revealed important associations that would have never been suspected based on prior knowledge or assumptions. To date, the discovered SNPs associated with complex human traits tend to have small effect sizes, requiring very large sample sizes to achieve robust statistical power. To address these issues, a number of efficient strategies have emerged for conducting GWAS, including combining study results across multiple studies using meta-analysis, collecting cases through electronic health records, and using samples collected from other studies as controls that have already been genotyped and made publicly available (e.g., through deposition of de-identified data into dbGaP or EGA). In certain scenarios, it may be attractive to use already genotyped controls and divert resources to standardized collection, phenotyping, and genotyping of cases only. This strategy, however, requires that careful attention be paid to the choice of "public controls" and to the comparability of genetic data between cases and the public controls to ensure that any allele frequency differences observed between groups is attributable to locus-specific effects rather than to a systematic bias due to poor matching (population stratification) or differential genotype calling (batch effects). The goal of this paper is to describe some of the potential pitfalls in using previously genotyped control data. We focus on considerations related to the choice of control groups, the use of different genotyping platforms, and approaches to deal with population stratification when cases and controls are genotyped across different platforms.
全基因组关联研究(GWAS)广泛应用于使用基因分型阵列识别各种疾病的易感基因座,这些阵列可以检测基因组中的已知多态性。GWAS 的一个特别优势是它对特定基因组元素(例如基因的编码或调控区域)是无偏的,并且它揭示了重要的关联,这些关联是基于先前的知识或假设永远不会被怀疑的。迄今为止,与复杂人类特征相关的发现的 SNP 往往具有较小的效应大小,需要非常大的样本量才能实现稳健的统计功效。为了解决这些问题,已经出现了许多用于进行 GWAS 的有效策略,包括使用荟萃分析结合多个研究的结果、通过电子健康记录收集病例,以及使用已经进行基因分型并公开可用的其他研究的样本作为对照(例如,通过将去识别数据存入 dbGaP 或 EGA)。在某些情况下,使用已经基因分型的对照并将资源转移到仅对病例进行标准化收集、表型和基因分型可能会很有吸引力。然而,这种策略需要仔细注意“公共对照”的选择以及病例和公共对照之间遗传数据的可比性,以确保观察到的群体之间的任何等位基因频率差异归因于特定基因座的影响,而不是由于匹配不良(群体分层)或差异基因型调用(批次效应)引起的系统偏差。本文的目的是描述使用先前基因分型对照数据的一些潜在陷阱。我们专注于与对照组选择、使用不同基因分型平台以及在病例和对照在不同平台上进行基因分型时处理群体分层的方法相关的考虑因素。