van Iperen E P A, Hovingh G K, Asselbergs F W, Zwinderman A H
Durrer Center for Cardiovascular Research, Netherlands Heart Institute, Utrecht, The Netherlands.
Department of Clinical Epidemiology, Biostatistics and Bioinformatics, Academic Medical Center, Amsterdam, The Netherlands.
PLoS One. 2017 Feb 28;12(2):e0172082. doi: 10.1371/journal.pone.0172082. eCollection 2017.
In the past decade many Genome-wide Association Studies (GWAS) were performed that discovered new associations between single-nucleotide polymorphisms (SNPs) and various phenotypes. Imputation methods are widely used in GWAS. They facilitate the phenotype association with variants that are not directly genotyped. Imputation methods can also be used to combine and analyse data genotyped on different genotyping arrays. In this study we investigated the imputation quality and efficiency of two different approaches of combining GWAS data from different genotyping platforms. We investigated whether combining data from different platforms before the actual imputation performs better than combining the data from different platforms after imputation.
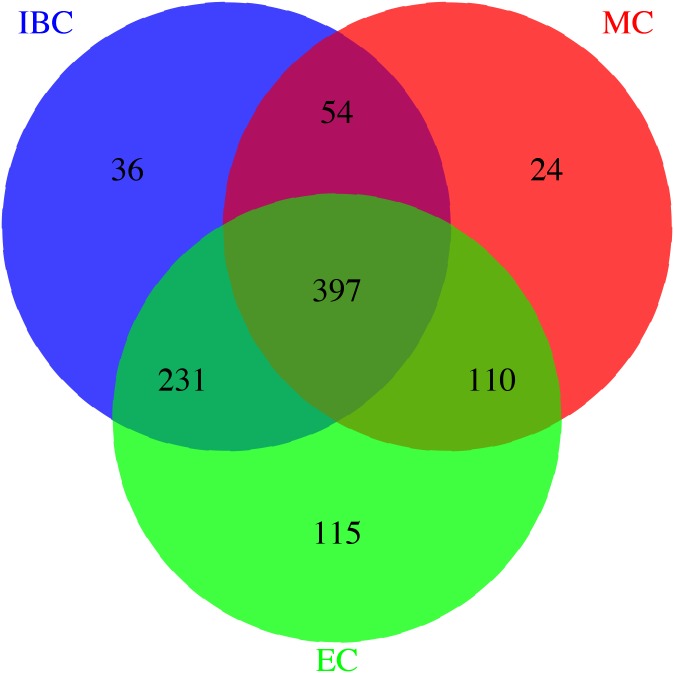
In total 979 unique individuals from the AMC-PAS cohort were genotyped on 3 different platforms. A total of 706 individuals were genotyped on the MetaboChip, a total of 757 individuals were genotyped on the 50K gene-centric Human CVD BeadChip, and a total of 955 individuals were genotyped on the HumanExome chip. A total of 397 individuals were genotyped on all 3 individual platforms. After pre-imputation quality control (QC), Minimac in combination with MaCH was used for the imputation of all samples with the 1,000 genomes reference panel. All imputed markers with an r2 value of <0.3 were excluded in our post-imputation QC.
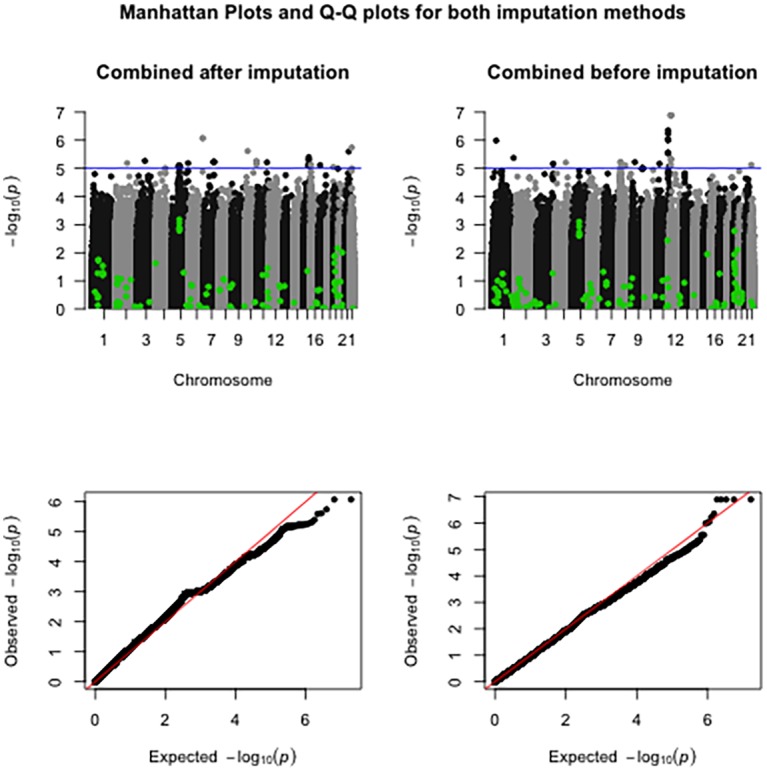
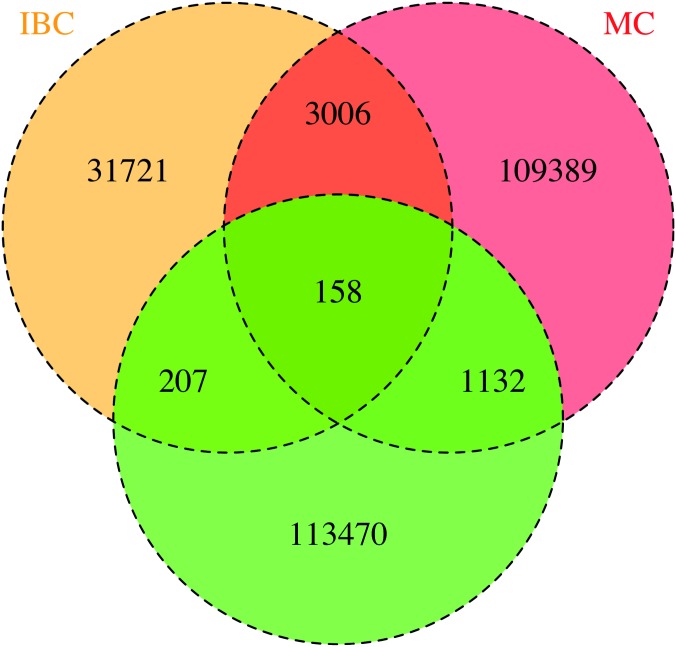
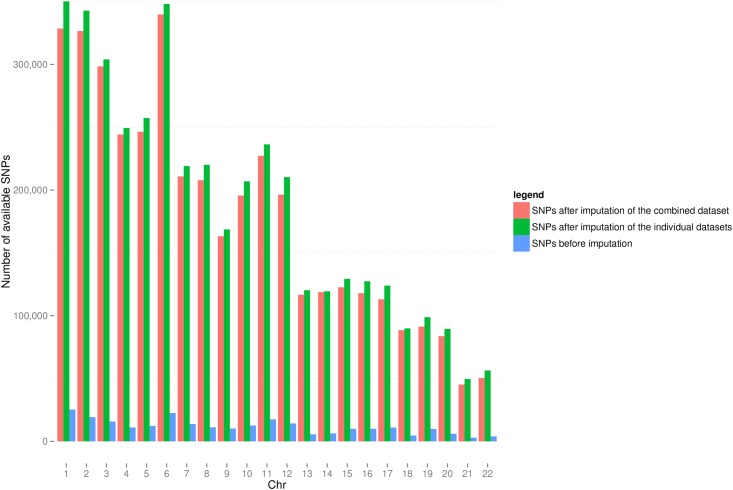
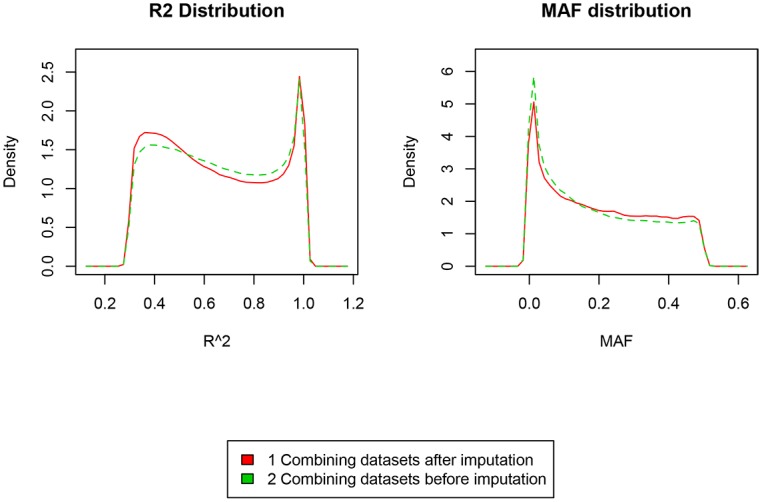
A total of 397 individuals were genotyped on all three platforms. All three datasets were carefully matched on strand, SNP ID and genomic coordinates. This resulted in a dataset of 979 unique individuals and a total of 258,925 unique markers. A total of 4,117,036 SNPs were available when imputation was performed before merging the three datasets. A total of 3,933,494 SNPs were available when imputation was done on the combined set. Our results suggest that imputation of individual datasets before merging performs slightly better than after combining the different datasets.
Imputation of datasets genotyped by different platforms before merging generates more SNPs than imputation after putting the datasets together.
在过去十年中,进行了许多全基因组关联研究(GWAS),发现了单核苷酸多态性(SNP)与各种表型之间的新关联。插补方法在GWAS中被广泛使用。它们有助于将表型与未直接进行基因分型的变异关联起来。插补方法还可用于合并和分析在不同基因分型阵列上进行基因分型的数据。在本研究中,我们调查了两种不同方法组合来自不同基因分型平台的GWAS数据的插补质量和效率。我们研究了在实际插补之前组合来自不同平台的数据是否比在插补之后组合来自不同平台的数据表现更好。
来自AMC-PAS队列的总共979名独特个体在3个不同平台上进行了基因分型。总共706名个体在代谢芯片上进行了基因分型,总共757名个体在以基因为中心的50K人类心血管疾病微珠芯片上进行了基因分型,总共955名个体在人类外显子芯片上进行了基因分型。总共397名个体在所有3个单独平台上进行了基因分型。在预插补质量控制(QC)之后,将Minimac与MaCH结合用于使用千人基因组参考面板对所有样本进行插补。在我们的插补后QC中,排除了r2值<0.3的所有插补标记。
总共397名个体在所有三个平台上进行了基因分型。所有三个数据集在链、SNP ID和基因组坐标上进行了仔细匹配。这产生了一个包含979名独特个体和总共258,925个独特标记的数据集。在合并三个数据集之前进行插补时,共有4,117,036个SNP可用。在合并集上进行插补时,共有3,933,494个SNP可用。我们的结果表明,在合并之前对各个数据集进行插补比在合并不同数据集之后进行插补表现略好。
在合并之前对由不同平台进行基因分型的数据集进行插补比将数据集放在一起之后进行插补产生更多的SNP。