Cross Sally H, Macalinao Danilo G, McKie Lisa, Rose Lorraine, Kearney Alison L, Rainger Joe, Thaung Caroline, Keighren Margaret, Jadeja Shalini, West Katrine, Kneeland Stephen C, Smith Richard S, Howell Gareth R, Young Fiona, Robertson Morag, van T' Hof Rob, John Simon W M, Jackson Ian J
MRC Human Genetics Unit, MRC IGMM, University of Edinburgh, Edinburgh, United Kingdom.
The Howard Hughes Medical Institute, The Jackson Laboratory, Bar Harbor, Maine, United States of America.
PLoS Genet. 2014 May 8;10(5):e1004359. doi: 10.1371/journal.pgen.1004359. eCollection 2014 May.
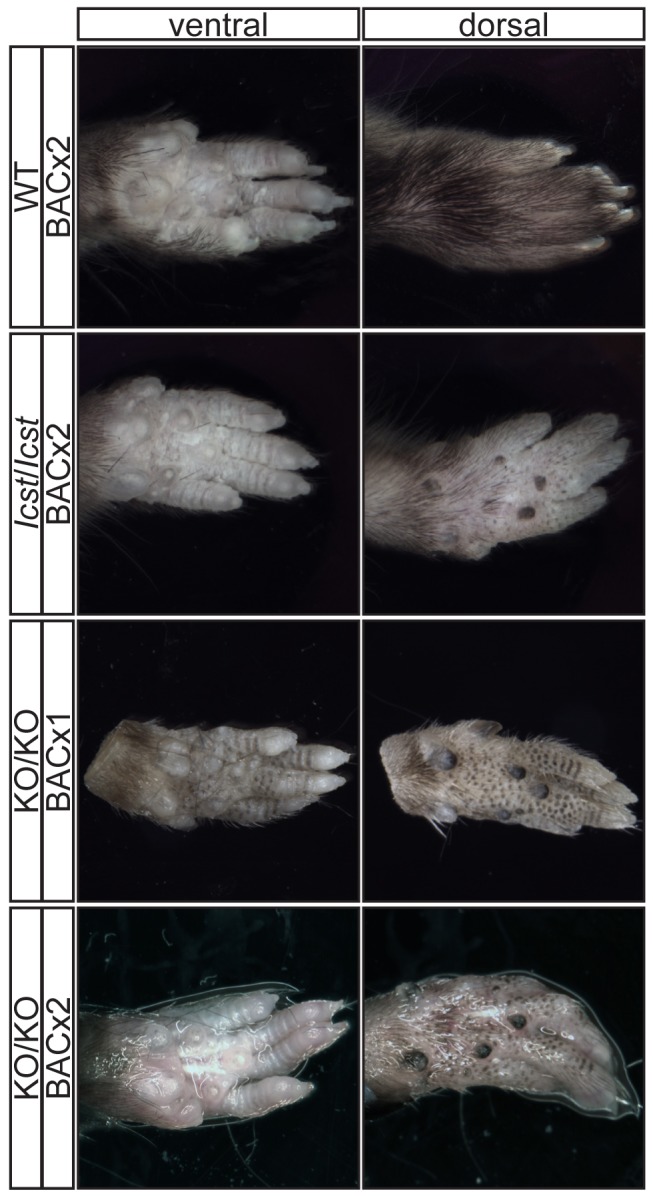
Mutations in the LIM-homeodomain transcription factor LMX1B cause nail-patella syndrome, an autosomal dominant pleiotrophic human disorder in which nail, patella and elbow dysplasia is associated with other skeletal abnormalities and variably nephropathy and glaucoma. It is thought to be a haploinsufficient disorder. Studies in the mouse have shown that during development Lmx1b controls limb dorsal-ventral patterning and is also required for kidney and eye development, midbrain-hindbrain boundary establishment and the specification of specific neuronal subtypes. Mice completely deficient for Lmx1b die at birth. In contrast to the situation in humans, heterozygous null mice do not have a mutant phenotype. Here we report a novel mouse mutant Icst, an N-ethyl-N-nitrosourea-induced missense substitution, V265D, in the homeodomain of LMX1B that abolishes DNA binding and thereby the ability to transactivate other genes. Although the homozygous phenotypic consequences of Icst and the null allele of Lmx1b are the same, heterozygous Icst elicits a phenotype whilst the null allele does not. Heterozygous Icst causes glaucomatous eye defects and is semi-lethal, probably due to kidney failure. We show that the null phenotype is rescued more effectively by an Lmx1b transgene than is Icst. Co-immunoprecipitation experiments show that both wild-type and Icst LMX1B are found in complexes with LIM domain binding protein 1 (LDB1), resulting in lower levels of functional LMX1B in Icst heterozygotes than null heterozygotes. We conclude that Icst is a dominant-negative allele of Lmx1b. These findings indicate a reassessment of whether nail-patella syndrome is always haploinsufficient. Furthermore, Icst is a rare example of a model of human glaucoma caused by mutation of the same gene in humans and mice.
LIM 同源结构域转录因子 LMX1B 的突变会导致指甲-髌骨综合征,这是一种常染色体显性多效性人类疾病,其中指甲、髌骨和肘部发育异常与其他骨骼异常以及不同程度的肾病和青光眼相关。它被认为是一种单倍剂量不足的疾病。对小鼠的研究表明,在发育过程中,Lmx1b 控制肢体背腹模式,也是肾脏和眼睛发育、中脑-后脑边界建立以及特定神经元亚型特化所必需的。完全缺乏 Lmx1b 的小鼠在出生时死亡。与人类的情况不同,杂合缺失小鼠没有突变表型。在此,我们报告一种新型小鼠突变体 Icst,它是由 N-乙基-N-亚硝基脲诱导的 LMX1B 同源结构域中的错义替代 V265D,该替代消除了 DNA 结合能力,从而丧失了反式激活其他基因的能力。尽管 Icst 的纯合表型后果与 Lmx1b 的无效等位基因相同,但杂合 Icst 会引发表型,而无效等位基因则不会。杂合 Icst 会导致青光眼性眼缺陷,并且可能由于肾衰竭而具有半致死性。我们表明,Lmx1b 转基因比 Icst 更有效地挽救了无效表型。免疫共沉淀实验表明,野生型和 Icst LMX1B 都与 LIM 结构域结合蛋白 1(LDB1)形成复合物,导致 Icst 杂合子中功能性 LMX1B 的水平低于无效杂合子。我们得出结论,Icst 是 Lmx1b 的显性负等位基因。这些发现表明需要重新评估指甲-髌骨综合征是否总是单倍剂量不足。此外,Icst 是人类和小鼠中由相同基因突变引起的人类青光眼模型的罕见例子。