Seth Sohan, Välimäki Niko, Kaski Samuel, Honkela Antti
Helsinki Institute for Information Technology HIIT, Department of Information and Computer Science, Aalto University, Espoo, Finland, Genome-Scale Biology Program and Department of Medical Genetics, University of Helsinki, Helsinki, Finland, and Helsinki Institute for Information Technology HIIT, Department of Computer Science, University of Helsinki, Helsinki, Finland.
Helsinki Institute for Information Technology HIIT, Department of Information and Computer Science, Aalto University, Espoo, Finland, Genome-Scale Biology Program and Department of Medical Genetics, University of Helsinki, Helsinki, Finland, and Helsinki Institute for Information Technology HIIT, Department of Computer Science, University of Helsinki, Helsinki, Finland Helsinki Institute for Information Technology HIIT, Department of Information and Computer Science, Aalto University, Espoo, Finland, Genome-Scale Biology Program and Department of Medical Genetics, University of Helsinki, Helsinki, Finland, and Helsinki Institute for Information Technology HIIT, Department of Computer Science, University of Helsinki, Helsinki, Finland.
Bioinformatics. 2014 Sep 1;30(17):2471-9. doi: 10.1093/bioinformatics/btu340. Epub 2014 May 19.
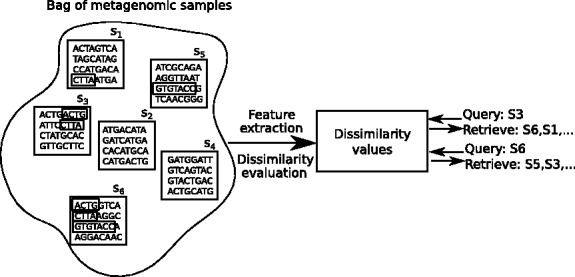
Over the recent years, the field of whole-metagenome shotgun sequencing has witnessed significant growth owing to the high-throughput sequencing technologies that allow sequencing genomic samples cheaper, faster and with better coverage than before. This technical advancement has initiated the trend of sequencing multiple samples in different conditions or environments to explore the similarities and dissimilarities of the microbial communities. Examples include the human microbiome project and various studies of the human intestinal tract. With the availability of ever larger databases of such measurements, finding samples similar to a given query sample is becoming a central operation.
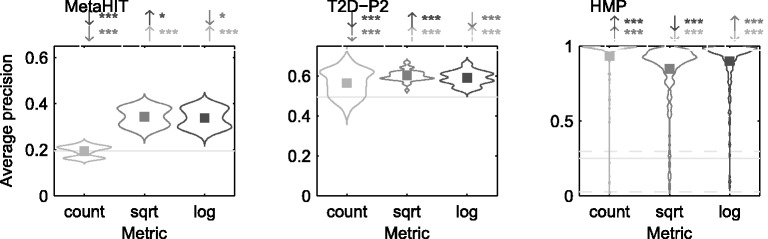
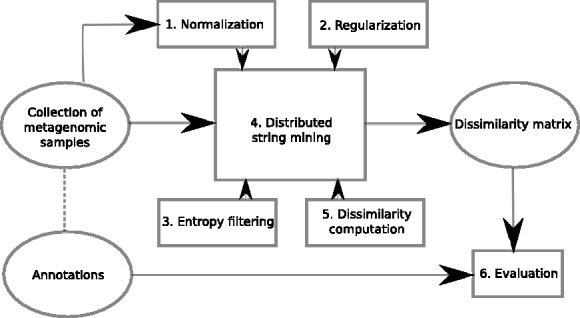
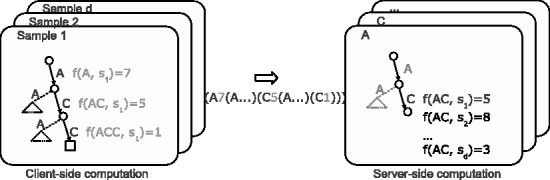
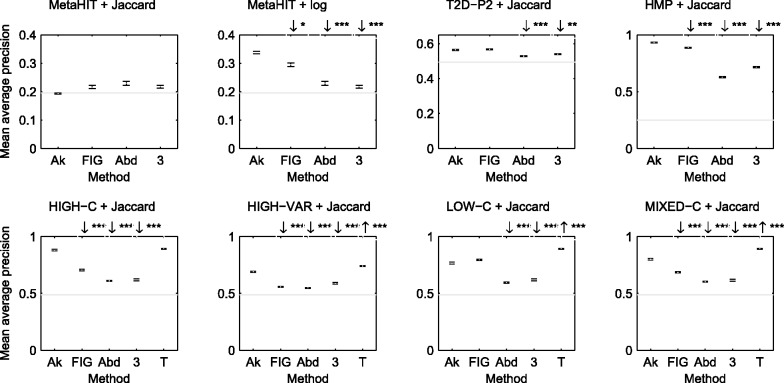
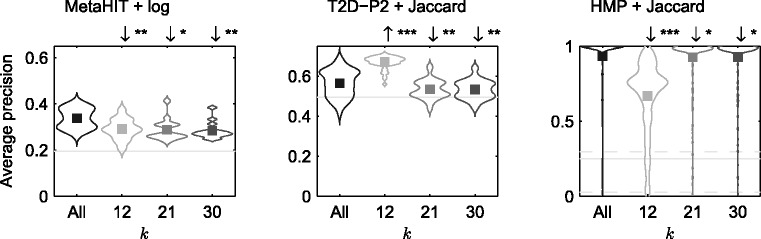
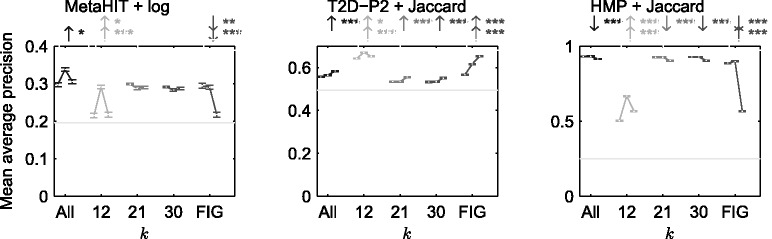
In this article, we develop a content-based exploration and retrieval method for whole-metagenome sequencing samples. We apply a distributed string mining framework to efficiently extract all informative sequence k-mers from a pool of metagenomic samples and use them to measure the dissimilarity between two samples. We evaluate the performance of the proposed approach on two human gut metagenome datasets as well as human microbiome project metagenomic samples. We observe significant enrichment for diseased gut samples in results of queries with another diseased sample and high accuracy in discriminating between different body sites even though the method is unsupervised.
A software implementation of the DSM framework is available at https://github.com/HIITMetagenomics/dsm-framework.
Supplementary data are available at Bioinformatics online.
近年来,由于高通量测序技术的发展,全基因组鸟枪法测序领域取得了显著进展。这些技术使得对基因组样本进行测序比以往更便宜、更快且覆盖度更高。这一技术进步引发了在不同条件或环境下对多个样本进行测序的趋势,以探索微生物群落的异同。例如人类微生物组计划以及对人类肠道的各种研究。随着此类测量的数据库不断增大,寻找与给定查询样本相似的样本正成为一项核心操作。
在本文中,我们开发了一种基于内容的全基因组测序样本探索与检索方法。我们应用分布式字符串挖掘框架,从宏基因组样本池中高效提取所有信息丰富的序列k-mer,并使用它们来衡量两个样本之间的差异。我们在两个人类肠道宏基因组数据集以及人类微生物组计划宏基因组样本上评估了所提出方法的性能。我们观察到,在使用另一个患病样本进行查询时,患病肠道样本在结果中显著富集,并且即使该方法是无监督的,在区分不同身体部位时也具有很高的准确性。
DSM框架的软件实现可在https://github.com/HIITMetagenomics/dsm-framework获取。
补充数据可在《生物信息学》在线获取。