Scientific Computing Department, STFC Daresbury Laboratory, Warrington, UK.
IBM Research, The Hartree Centre, Warrington, UK.
Microbiome. 2019 Mar 16;7(1):40. doi: 10.1186/s40168-019-0653-2.
The growth in publically available microbiome data in recent years has yielded an invaluable resource for genomic research, allowing for the design of new studies, augmentation of novel datasets and reanalysis of published works. This vast amount of microbiome data, as well as the widespread proliferation of microbiome research and the looming era of clinical metagenomics, means there is an urgent need to develop analytics that can process huge amounts of data in a short amount of time. To address this need, we propose a new method for tyrhe compact representation of microbiome sequencing data using similarity-preserving sketches of streaming k-mer spectra. These sketches allow for dissimilarity estimation, rapid microbiome catalogue searching and classification of microbiome samples in near real time.
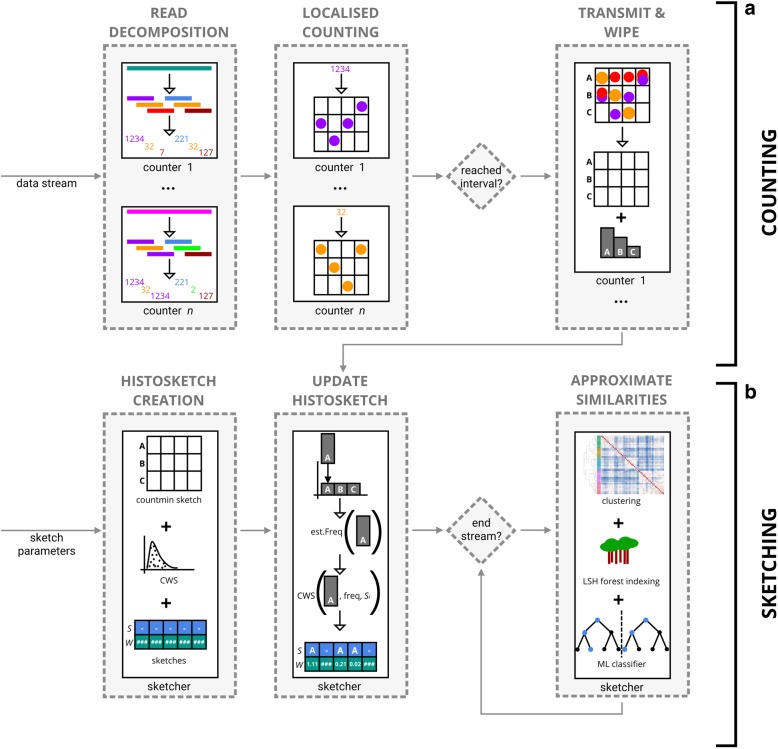
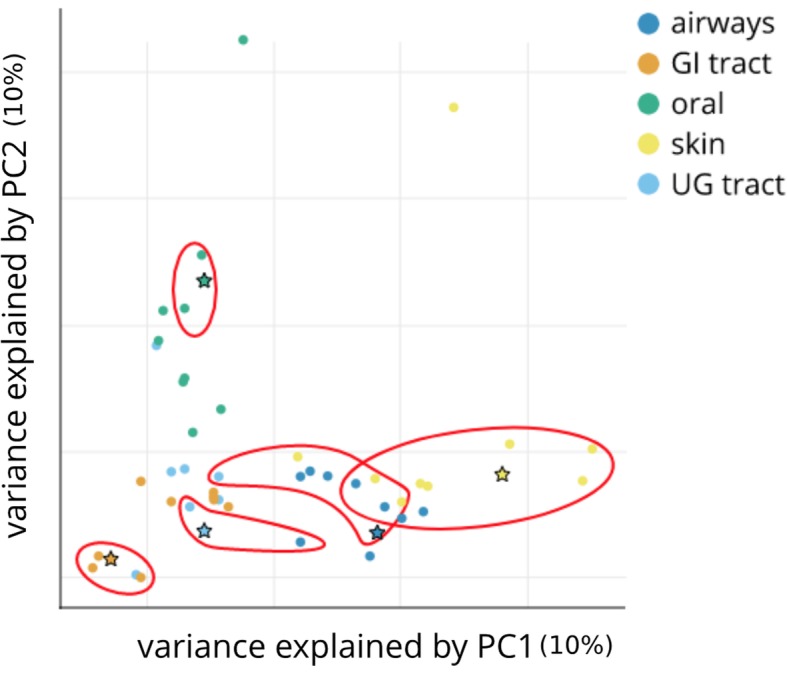
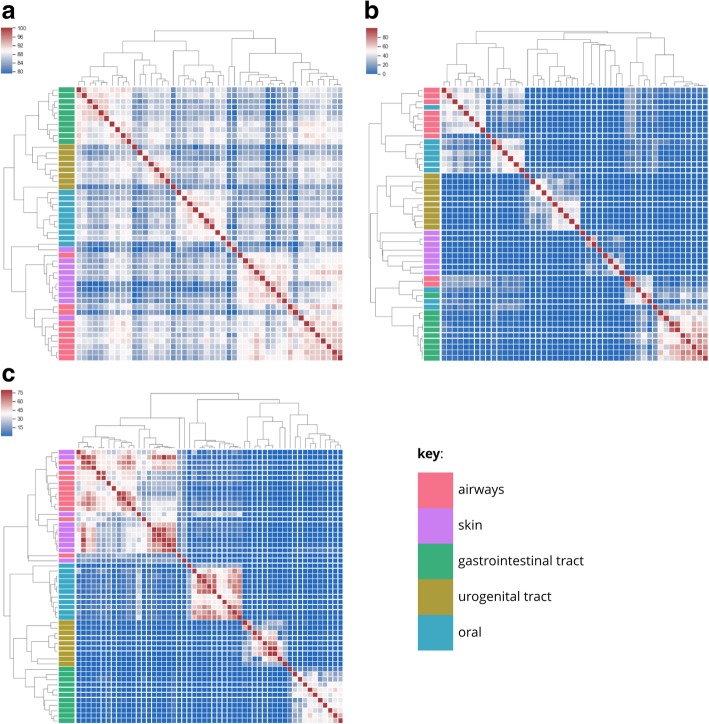
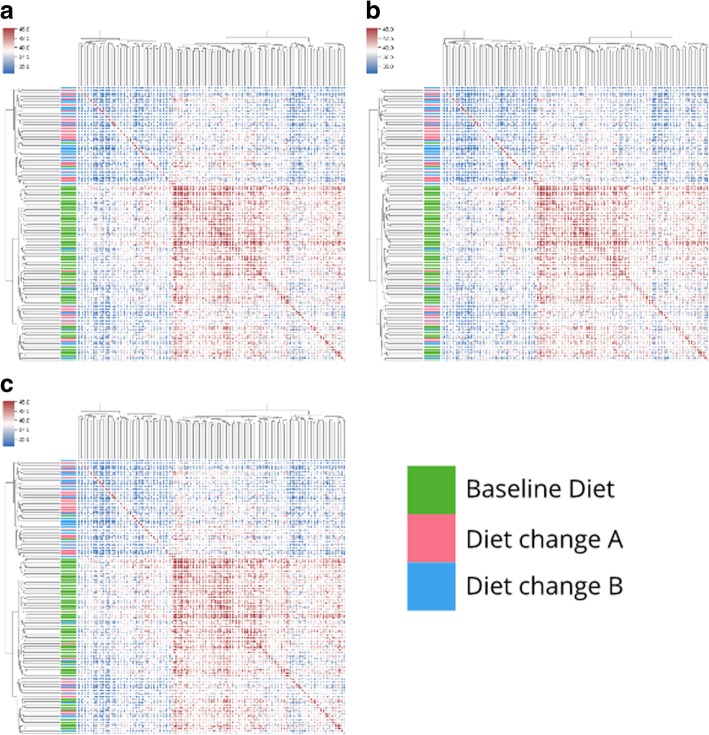
We apply streaming histogram sketching to microbiome samples as a form of dimensionality reduction, creating a compressed 'histosketch' that can efficiently represent microbiome k-mer spectra. Using public microbiome datasets, we show that histosketches can be clustered by sample type using the pairwise Jaccard similarity estimation, consequently allowing for rapid microbiome similarity searches via a locality sensitive hashing indexing scheme. Furthermore, we use a 'real life' example to show that histosketches can train machine learning classifiers to accurately label microbiome samples. Specifically, using a collection of 108 novel microbiome samples from a cohort of premature neonates, we trained and tested a random forest classifier that could accurately predict whether the neonate had received antibiotic treatment (97% accuracy, 96% precision) and could subsequently be used to classify microbiome data streams in less than 3 s.
Our method offers a new approach to rapidly process microbiome data streams, allowing samples to be rapidly clustered, indexed and classified. We also provide our implementation, Histosketching Using Little K-mers (HULK), which can histosketch a typical 2 GB microbiome in 50 s on a standard laptop using four cores, with the sketch occupying 3000 bytes of disk space. ( https://github.com/will-rowe/hulk ).
近年来,公共微生物组数据的增长为基因组研究提供了宝贵的资源,使得能够设计新的研究,增加新的数据集,并重新分析已发表的工作。大量的微生物组数据,以及微生物组研究的广泛普及和临床宏基因组学的即将到来,意味着迫切需要开发能够在短时间内处理大量数据的分析工具。为了满足这一需求,我们提出了一种使用流 k-mer 谱相似性保留草图对微生物组测序数据进行紧凑表示的新方法。这些草图允许进行不相似性估计、快速微生物组目录搜索和微生物组样本的分类,几乎可以实时进行。
我们将流直方图草图应用于微生物组样本作为一种降维形式,创建了一个可以有效表示微生物组 k-mer 谱的压缩“histosketch”。使用公共微生物组数据集,我们表明可以使用样本类型的成对 Jaccard 相似性估计对 histosketches 进行聚类,从而可以通过局部敏感哈希索引方案快速进行微生物组相似性搜索。此外,我们使用一个“现实生活”的例子来说明 histosketches 可以训练机器学习分类器来准确标记微生物组样本。具体来说,使用来自早产儿队列的 108 个新型微生物组样本的集合,我们训练并测试了一个随机森林分类器,该分类器可以准确预测新生儿是否接受了抗生素治疗(准确率为 97%,精度为 96%),并且可以随后用于在不到 3 秒的时间内对微生物组数据流进行分类。
我们的方法为快速处理微生物组数据流提供了一种新方法,允许快速对样本进行聚类、索引和分类。我们还提供了我们的实现,即使用小 k-mer 的 Histosketching(HULK),它可以在标准笔记本电脑上使用四个核在 50 秒内对典型的 2GB 微生物组进行 histosketching,草图占用 3000 字节的磁盘空间。(https://github.com/will-rowe/hulk)