1] Center for Genome Sciences and Systems Biology, Washington University School of Medicine, St Louis, Missouri 63108, USA [2].
1] Center for Genome Sciences and Systems Biology, Washington University School of Medicine, St Louis, Missouri 63108, USA [2] Department of Pathology and Immunology, Washington University School of Medicine, St Louis, Missouri 63110, USA [3].
Nature. 2014 May 29;509(7502):612-6. doi: 10.1038/nature13377. Epub 2014 May 21.
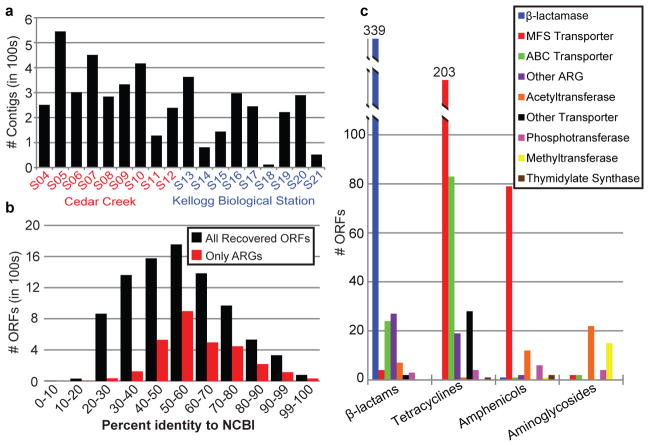
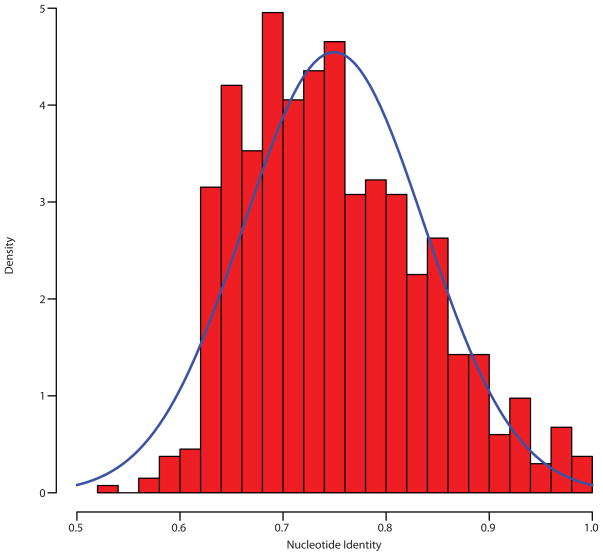
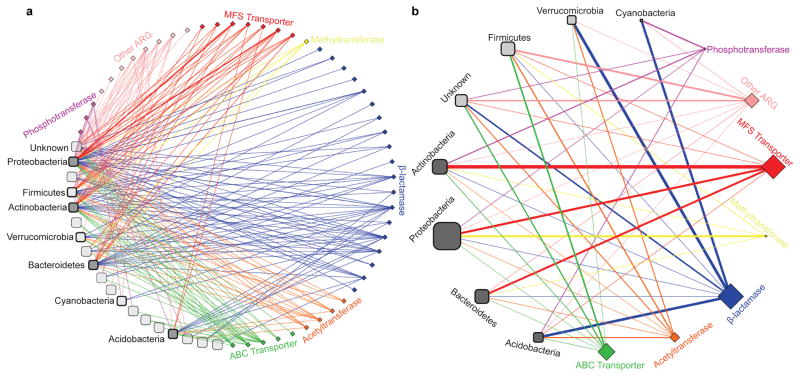
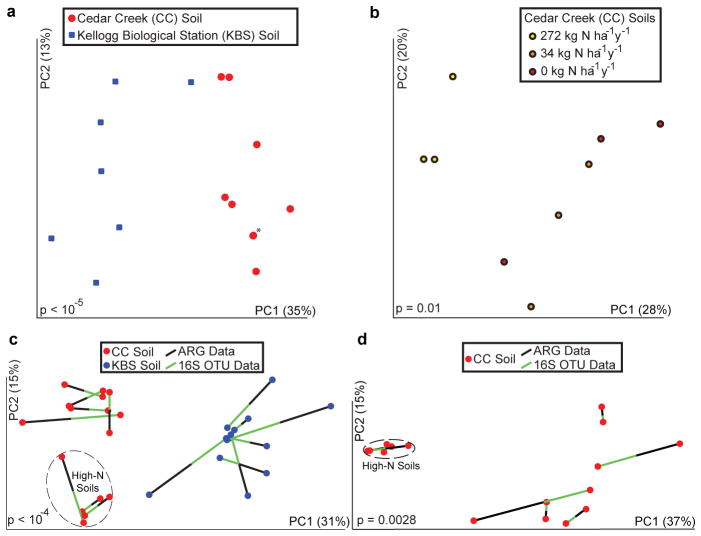
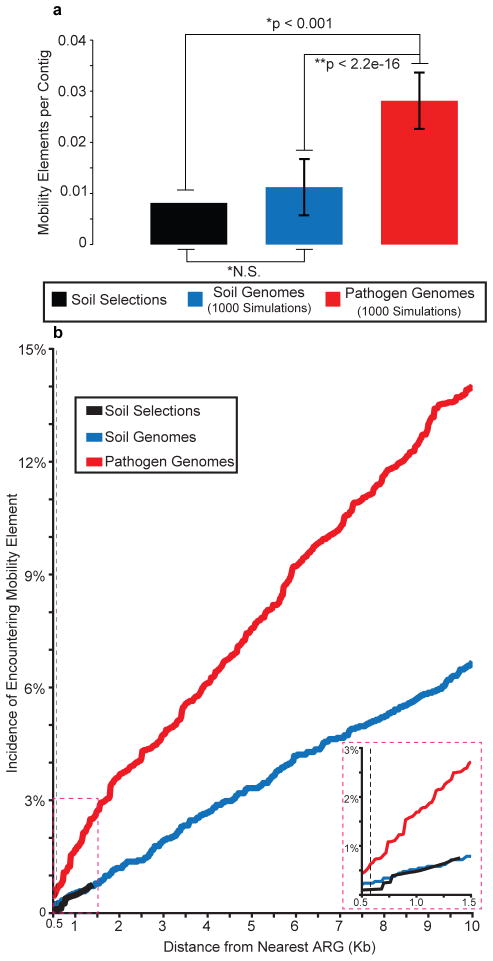
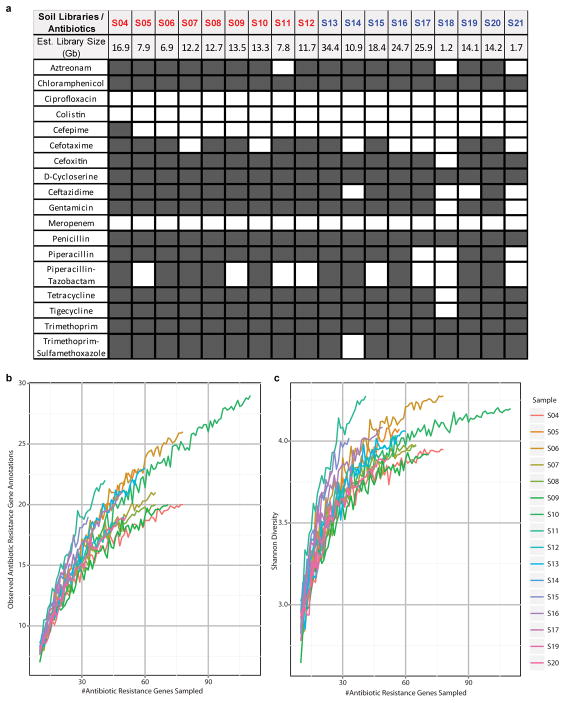
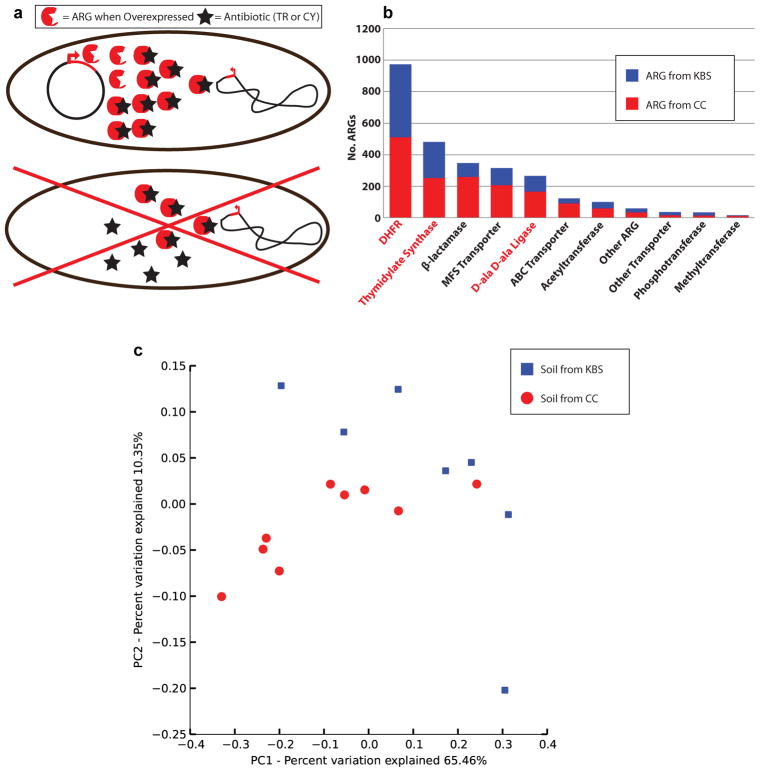
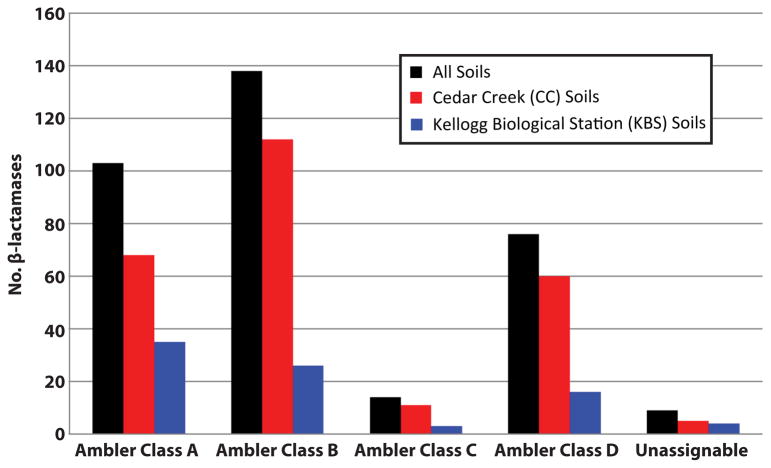
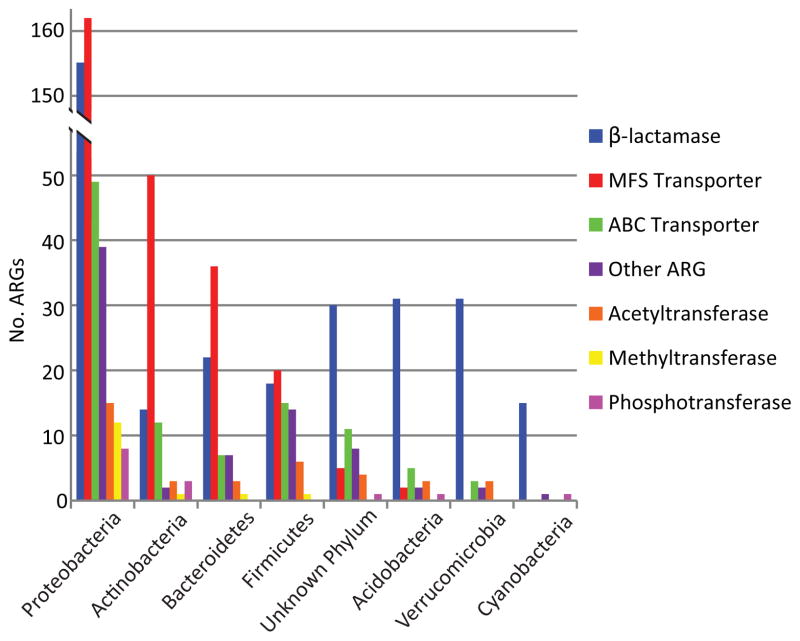
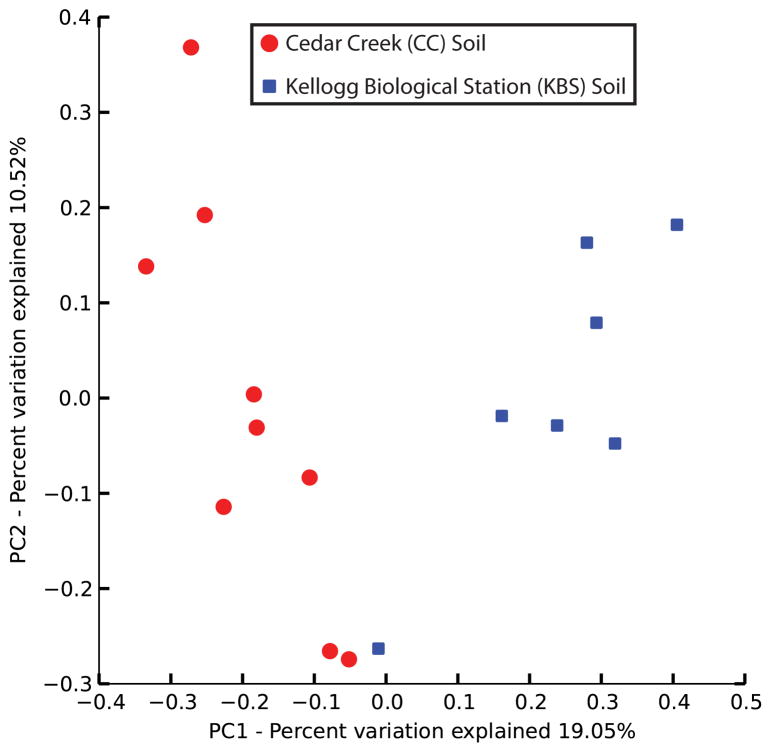
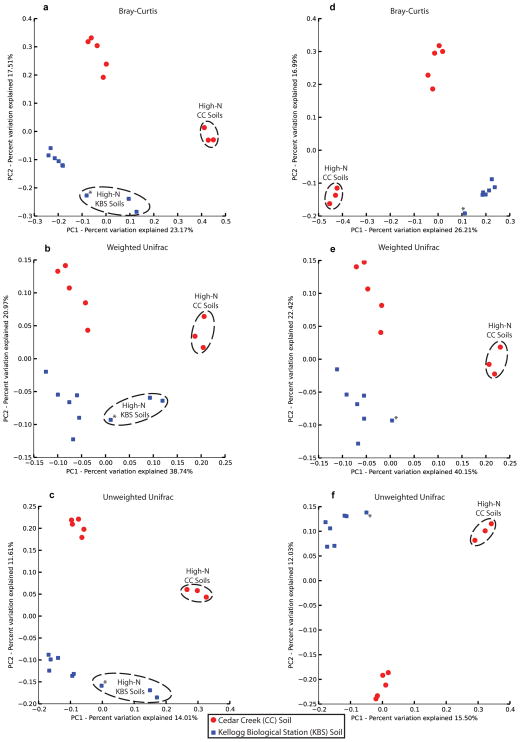
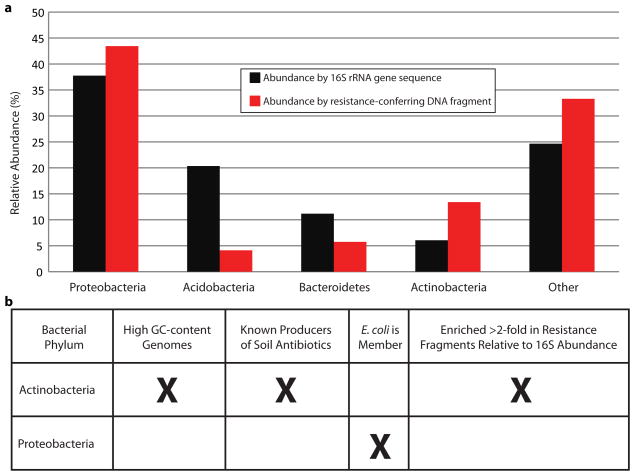
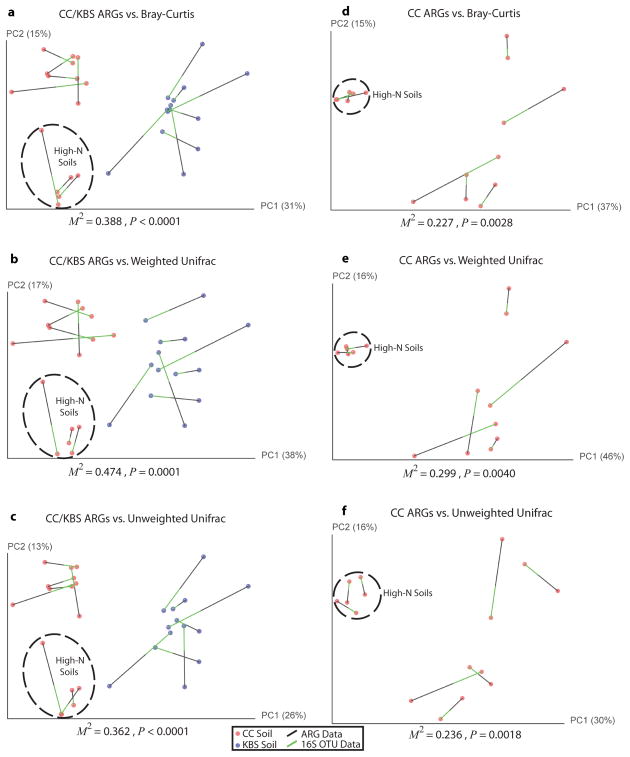
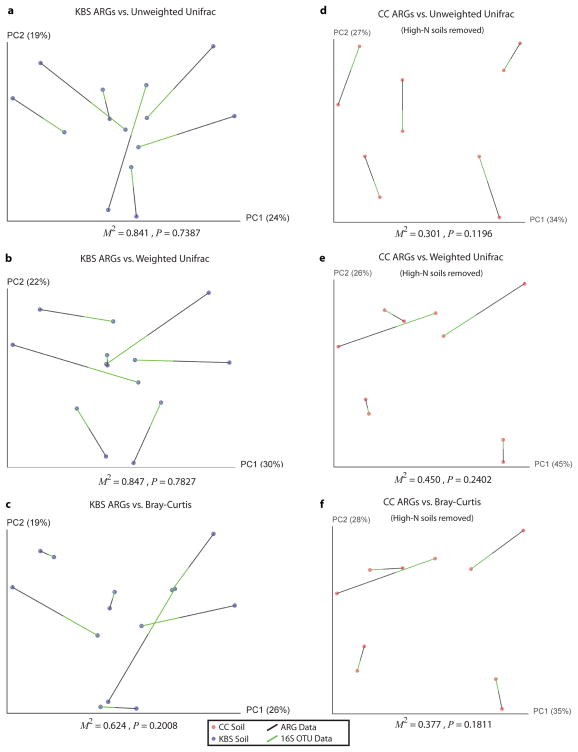
Ancient and diverse antibiotic resistance genes (ARGs) have previously been identified from soil, including genes identical to those in human pathogens. Despite the apparent overlap between soil and clinical resistomes, factors influencing ARG composition in soil and their movement between genomes and habitats remain largely unknown. General metagenome functions often correlate with the underlying structure of bacterial communities. However, ARGs are proposed to be highly mobile, prompting speculation that resistomes may not correlate with phylogenetic signatures or ecological divisions. To investigate these relationships, we performed functional metagenomic selections for resistance to 18 antibiotics from 18 agricultural and grassland soils. The 2,895 ARGs we discovered were mostly new, and represent all major resistance mechanisms. We demonstrate that distinct soil types harbour distinct resistomes, and that the addition of nitrogen fertilizer strongly influenced soil ARG content. Resistome composition also correlated with microbial phylogenetic and taxonomic structure, both across and within soil types. Consistent with this strong correlation, mobility elements (genes responsible for horizontal gene transfer between bacteria such as transposases and integrases) syntenic with ARGs were rare in soil by comparison with sequenced pathogens, suggesting that ARGs may not transfer between soil bacteria as readily as is observed between human pathogens. Together, our results indicate that bacterial community composition is the primary determinant of soil ARG content, challenging previous hypotheses that horizontal gene transfer effectively decouples resistomes from phylogeny.
先前已从土壤中鉴定出古老且多样化的抗生素耐药基因 (ARGs),其中包括与人类病原体中相同的基因。尽管土壤和临床耐药组之间明显存在重叠,但影响土壤中 ARG 组成及其在基因组和生境之间转移的因素在很大程度上仍不清楚。一般宏基因组功能通常与细菌群落的基本结构相关。然而,ARGs 被认为具有高度的可移动性,这促使人们推测耐药组可能与系统发育特征或生态分区无关。为了研究这些关系,我们从 18 个农业和草原土壤中进行了针对 18 种抗生素的抗性功能宏基因组选择。我们发现的 2895 个 ARG 主要是新的,代表了所有主要的耐药机制。我们证明不同的土壤类型具有不同的耐药组,并且添加氮肥强烈影响土壤 ARG 含量。耐药组的组成也与微生物系统发育和分类结构相关,无论是在土壤类型之间还是在土壤类型内。与这种强烈相关性一致的是,与测序病原体相比,移动元件(负责细菌之间水平基因转移的基因,如转座酶和整合酶)与 ARG 之间的同线性在土壤中很少见,这表明 ARGs 可能不像在人类病原体之间那样容易在土壤细菌之间转移。总的来说,我们的结果表明细菌群落组成是土壤 ARG 含量的主要决定因素,这挑战了先前关于水平基因转移有效地将耐药组与系统发育解耦的假设。