D'Abramo Marco, Besker Neva, Chillemi Giovanni, Grottesi Alessandro
CINECA Rome, Italy ; Dipartimento di Chimica, Sapienza University of Rome Rome, Italy.
Dipartimento di Scienze e Tecnologie Chimiche, Università di Roma "Tor Vergata," Rome, Italy.
Front Genet. 2014 May 13;5:128. doi: 10.3389/fgene.2014.00128. eCollection 2014.
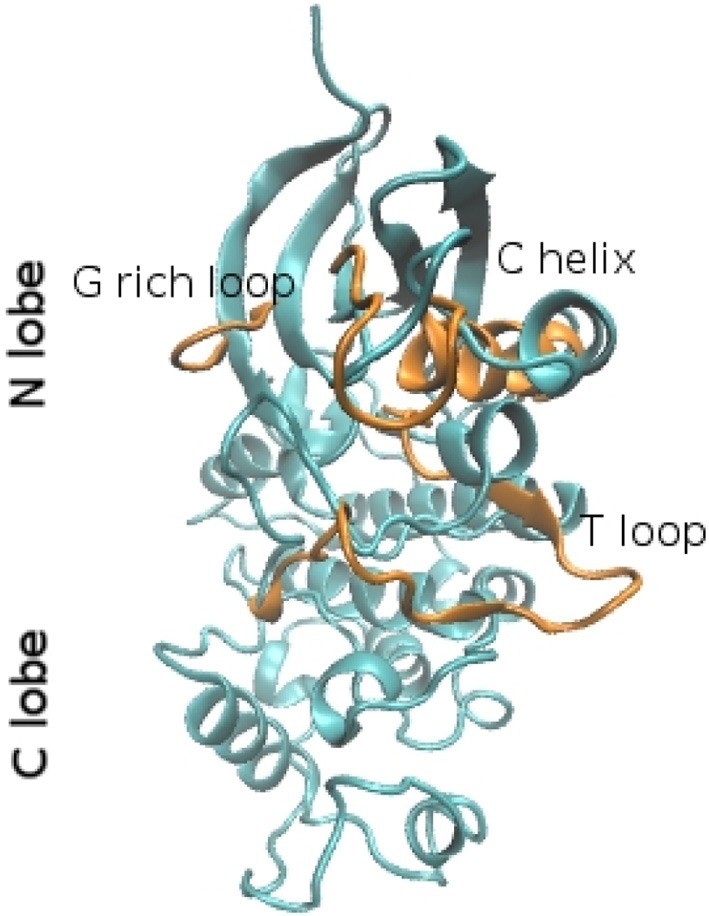
Protein kinases work because their flexibility allows to continuously switch from inactive to active form. Despite the large number of structures experimentally determined in such states, the mechanism of their conformational transitions as well as the transition pathways are not easily to capture. In this regard, computational methods can help to shed light on such an issue. However, due to the intrinsic sampling limitations, much efforts have been done to model in a realistic way the conformational changes occurring in protein kinases. In this review we will address the principal biological achievements and structural aspects in studying kinases conformational transitions and will focus on the main challenges related to computational approaches such as molecular modeling and MD simulations.
蛋白激酶能够发挥作用是因为其灵活性使其能够持续从无活性形式转变为活性形式。尽管已经通过实验确定了大量处于此类状态的结构,但其构象转变机制以及转变途径却不易捕捉。在这方面,计算方法有助于阐明此类问题。然而,由于固有的采样限制,人们已经付出了很多努力来以现实的方式模拟蛋白激酶中发生的构象变化。在本综述中,我们将阐述研究激酶构象转变的主要生物学成果和结构方面,并将重点关注与分子建模和分子动力学模拟等计算方法相关的主要挑战。