Kawakami Takeshi, Smeds Linnéa, Backström Niclas, Husby Arild, Qvarnström Anna, Mugal Carina F, Olason Pall, Ellegren Hans
Department of Evolutionary Biology, Evolutionary Biology Centre (EBC), Uppsala University, Norbyvägen 18D, SE-752 36, Uppsala, Sweden.
Mol Ecol. 2014 Aug;23(16):4035-58. doi: 10.1111/mec.12810. Epub 2014 Jun 17.
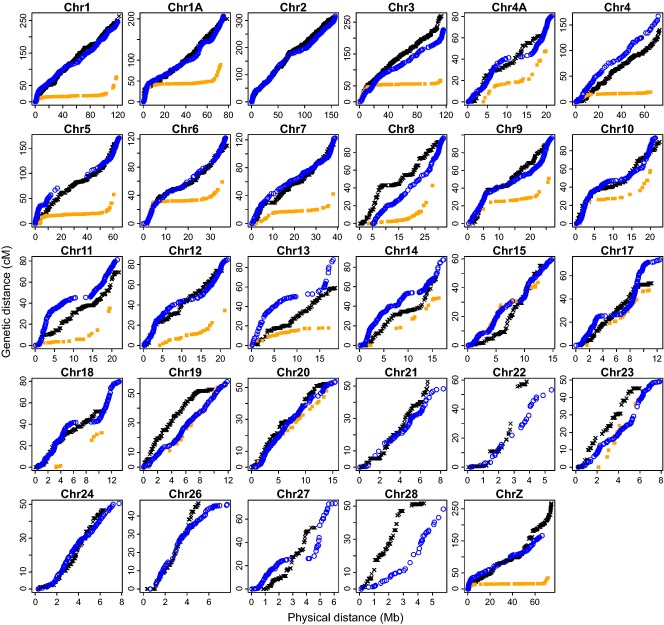
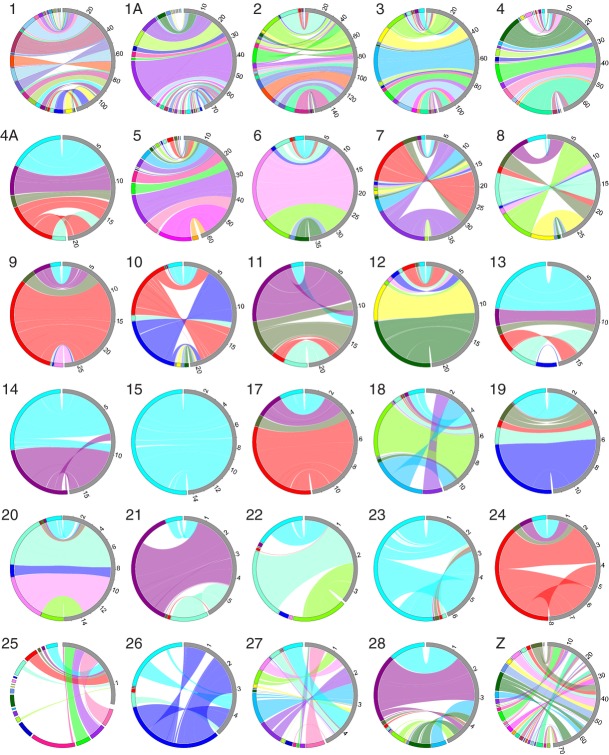
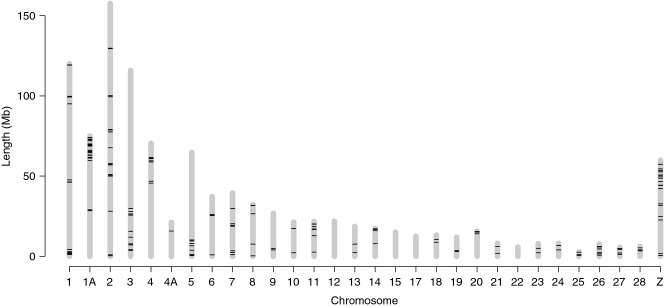
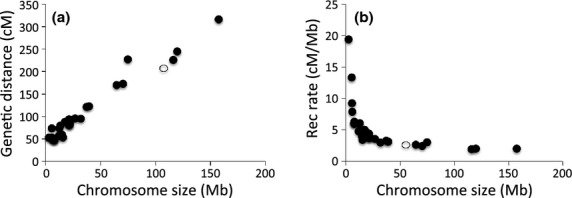
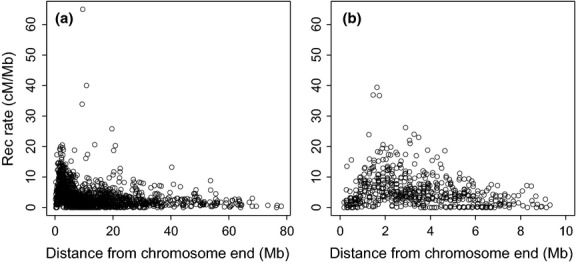
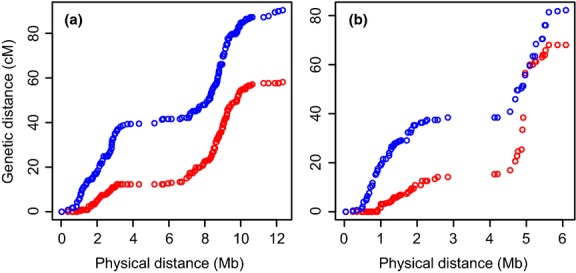
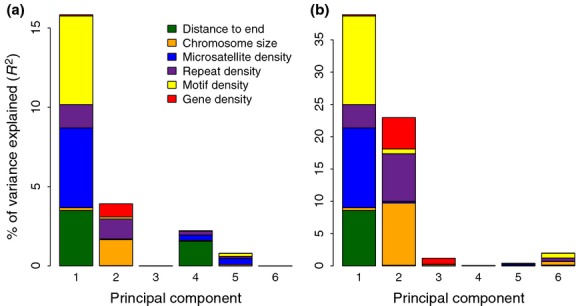
Detailed linkage and recombination rate maps are necessary to use the full potential of genome sequencing and population genomic analyses. We used a custom collared flycatcher 50 K SNP array to develop a high-density linkage map with 37 262 markers assigned to 34 linkage groups in 33 autosomes and the Z chromosome. The best-order map contained 4215 markers, with a total distance of 3132 cM and a mean genetic distance between markers of 0.12 cM. Facilitated by the array being designed to include markers from most scaffolds, we obtained a second-generation assembly of the flycatcher genome that approaches full chromosome sequences (N50 super-scaffold size 20.2 Mb and with 1.042 Gb (of 1.116 Gb) anchored to and mostly ordered and oriented along chromosomes). We found that flycatcher and zebra finch chromosomes are entirely syntenic but that inversions at mean rates of 1.5-2.0 event (6.6-7.5 Mb) per My have changed the organization within chromosomes, rates high enough for inversions to potentially have been involved with many speciation events during avian evolution. The mean recombination rate was 3.1 cM/Mb and correlated closely with chromosome size, from 2 cM/Mb for chromosomes >100 Mb to >10 cM/Mb for chromosomes <10 Mb. This size dependence seemed entirely due to an obligate recombination event per chromosome; if 50 cM was subtracted from the genetic lengths of chromosomes, the rate per physical unit DNA was constant across chromosomes. Flycatcher recombination rate showed similar variation along chromosomes as chicken but lacked the large interior recombination deserts characteristic of zebra finch chromosomes.
要充分发挥基因组测序和群体基因组分析的潜力,详细的连锁和重组率图谱是必不可少的。我们使用定制的白领姬鹟50K SNP阵列来构建高密度连锁图谱,将37262个标记分配到33条常染色体和Z染色体的34个连锁群中。最佳排序图谱包含4215个标记,总距离为3132厘摩,标记间平均遗传距离为0.12厘摩。由于该阵列设计为包含来自大多数支架的标记,我们获得了白领姬鹟基因组的第二代组装,其接近完整的染色体序列(N50超级支架大小为20.2 Mb,1.116 Gb中的1.042 Gb锚定在染色体上并大多按顺序排列和定向)。我们发现白领姬鹟和斑胸草雀的染色体完全是同线的,但平均每百万年1.5 - 2.0次事件(6.6 - 7.5 Mb)的倒位改变了染色体内的组织,这些速率高到足以使倒位可能参与了鸟类进化过程中的许多物种形成事件。平均重组率为3.1厘摩/兆碱基,与染色体大小密切相关,大于100 Mb的染色体为2厘摩/兆碱基,小于10 Mb的染色体大于10厘摩/兆碱基。这种大小依赖性似乎完全是由于每条染色体上有一次必然的重组事件;如果从染色体的遗传长度中减去50厘摩,那么每物理单位DNA的速率在各染色体间是恒定的。白领姬鹟的重组率沿染色体的变化与鸡相似,但缺乏斑胸草雀染色体特有的大的内部重组荒漠。