Chen Ying, McGee Jeremy, Chen Xianming, Doman Thompson N, Gong Xueqian, Zhang Youyan, Hamm Nicole, Ma Xiwen, Higgs Richard E, Bhagwat Shripad V, Buchanan Sean, Peng Sheng-Bin, Staschke Kirk A, Yadav Vipin, Yue Yong, Kouros-Mehr Hosein
Department of Oncology, Eli Lilly and Company, Indianapolis, Indiana, United States of America.
PLoS One. 2014 May 29;9(5):e98293. doi: 10.1371/journal.pone.0098293. eCollection 2014.
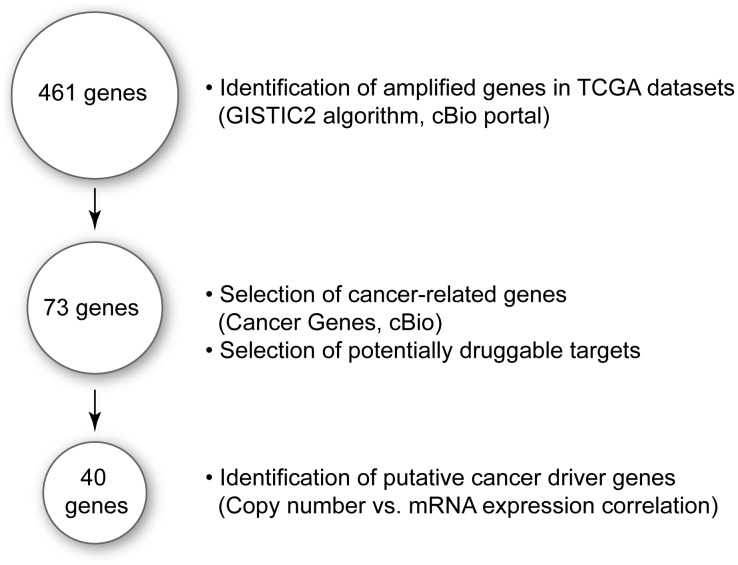
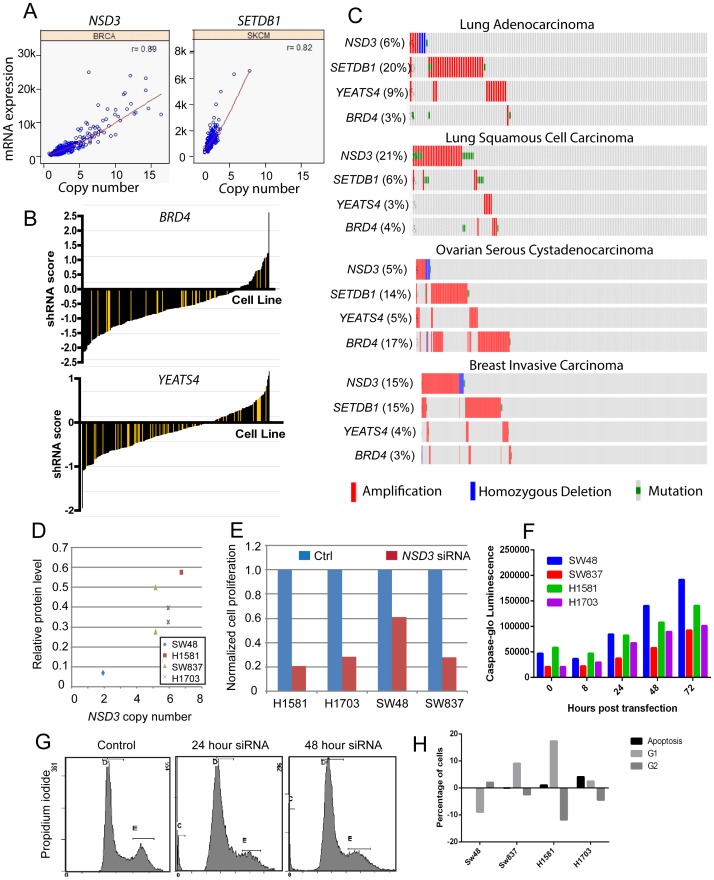
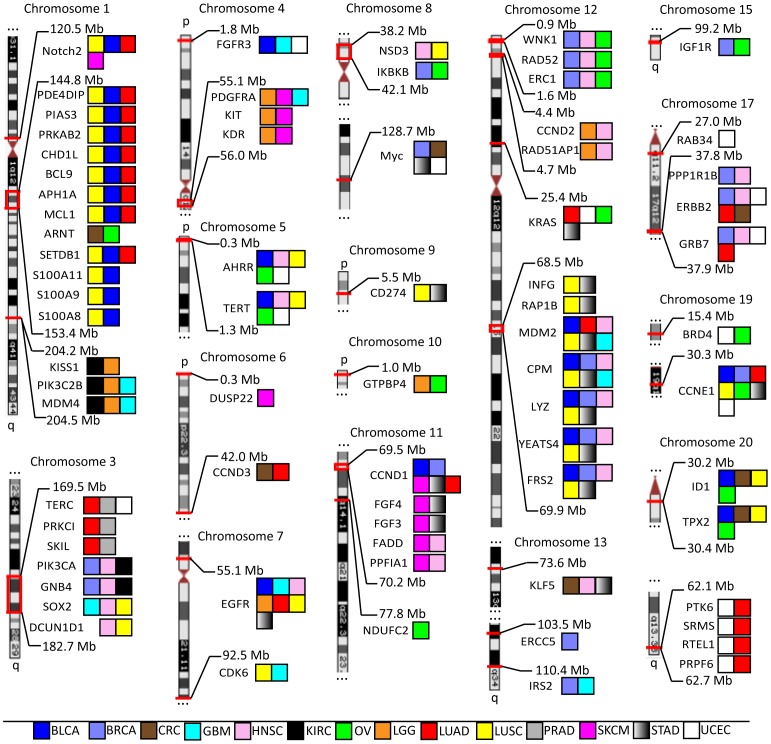
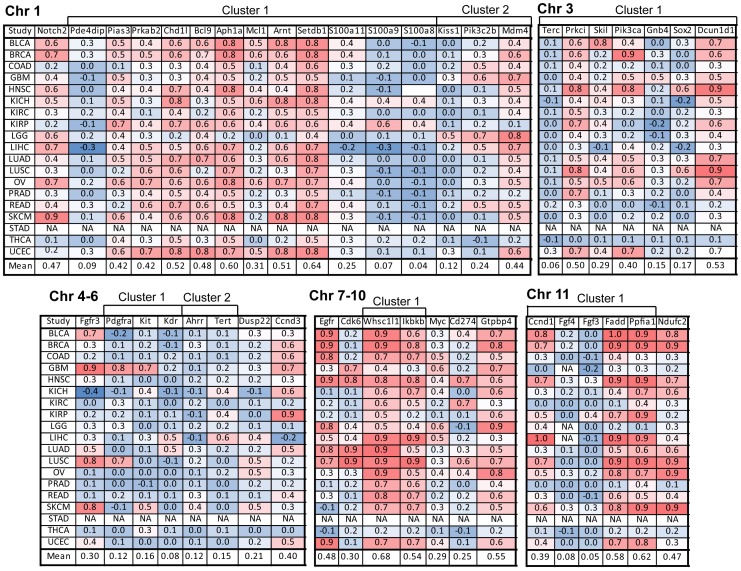
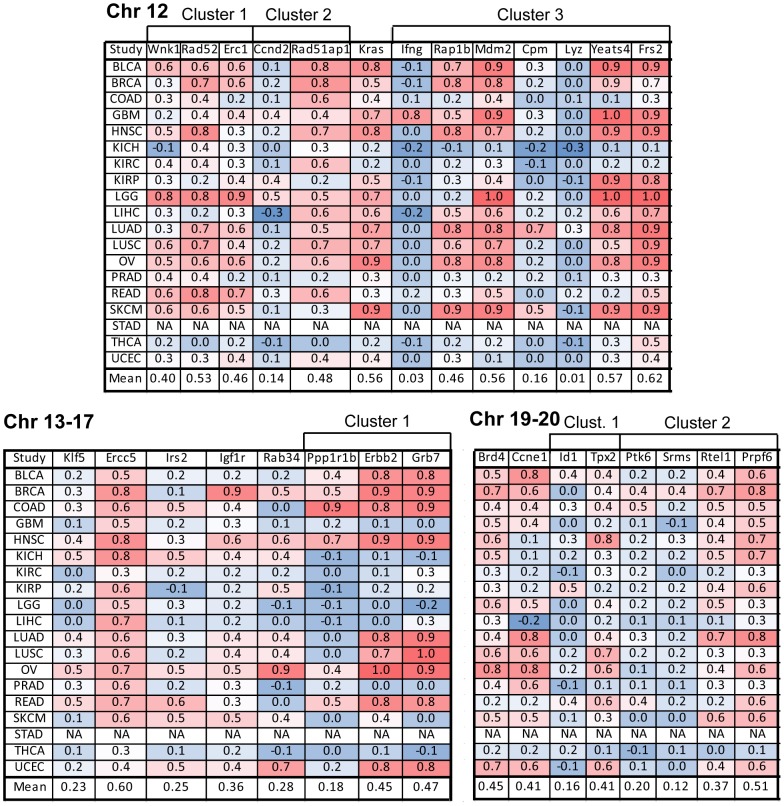
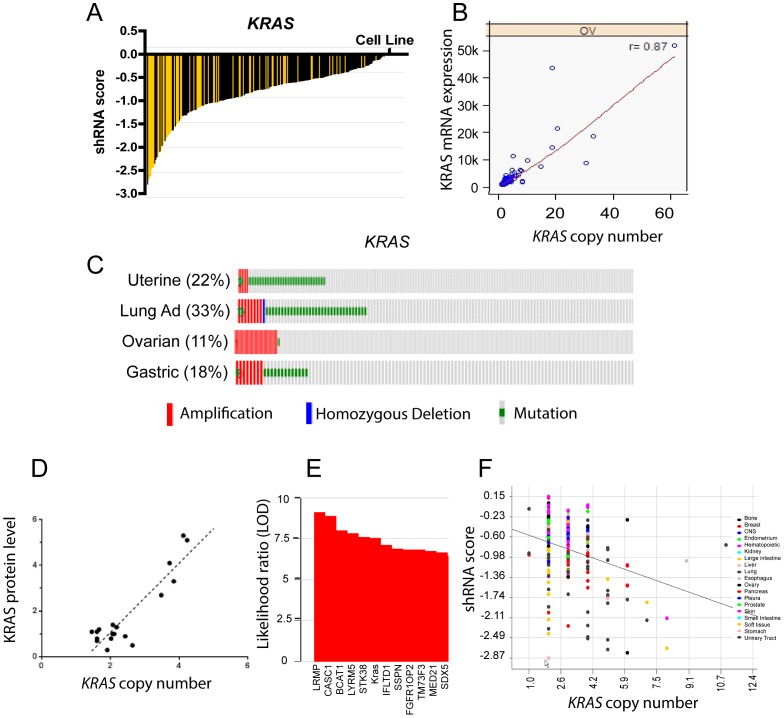
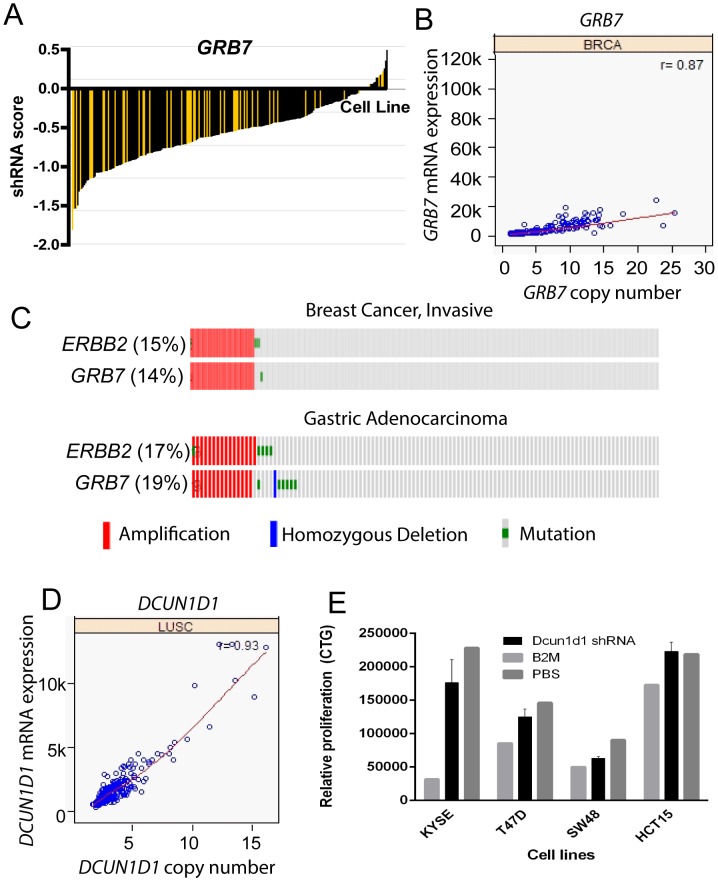
The Cancer Genome Atlas (TCGA) projects have advanced our understanding of the driver mutations, genetic backgrounds, and key pathways activated across cancer types. Analysis of TCGA datasets have mostly focused on somatic mutations and translocations, with less emphasis placed on gene amplifications. Here we describe a bioinformatics screening strategy to identify putative cancer driver genes amplified across TCGA datasets. We carried out GISTIC2 analysis of TCGA datasets spanning 16 cancer subtypes and identified 486 genes that were amplified in two or more datasets. The list was narrowed to 75 cancer-associated genes with potential "druggable" properties. The majority of the genes were localized to 14 amplicons spread across the genome. To identify potential cancer driver genes, we analyzed gene copy number and mRNA expression data from individual patient samples and identified 42 putative cancer driver genes linked to diverse oncogenic processes. Oncogenic activity was further validated by siRNA/shRNA knockdown and by referencing the Project Achilles datasets. The amplified genes represented a number of gene families, including epigenetic regulators, cell cycle-associated genes, DNA damage response/repair genes, metabolic regulators, and genes linked to the Wnt, Notch, Hedgehog, JAK/STAT, NF-KB and MAPK signaling pathways. Among the 42 putative driver genes were known driver genes, such as EGFR, ERBB2 and PIK3CA. Wild-type KRAS was amplified in several cancer types, and KRAS-amplified cancer cell lines were most sensitive to KRAS shRNA, suggesting that KRAS amplification was an independent oncogenic event. A number of MAP kinase adapters were co-amplified with their receptor tyrosine kinases, such as the FGFR adapter FRS2 and the EGFR family adapters GRB2 and GRB7. The ubiquitin-like ligase DCUN1D1 and the histone methyltransferase NSD3 were also identified as novel putative cancer driver genes. We discuss the patient tailoring implications for existing cancer drug targets and we further discuss potential novel opportunities for drug discovery efforts.
癌症基因组图谱(TCGA)项目增进了我们对驱动突变、遗传背景以及各类癌症中激活的关键信号通路的理解。对TCGA数据集的分析大多聚焦于体细胞突变和易位,而对基因扩增的关注较少。在此,我们描述了一种生物信息学筛选策略,以识别在TCGA数据集中扩增的假定癌症驱动基因。我们对涵盖16种癌症亚型的TCGA数据集进行了GISTIC2分析,鉴定出在两个或更多数据集中扩增的486个基因。该列表被缩减至75个具有潜在“可成药”特性的癌症相关基因。大多数基因定位于分布在基因组中的14个扩增子上。为了识别潜在的癌症驱动基因,我们分析了个体患者样本的基因拷贝数和mRNA表达数据,并鉴定出42个与多种致癌过程相关的假定癌症驱动基因。通过siRNA/shRNA敲低以及参考阿喀琉斯计划数据集进一步验证了致癌活性。扩增的基因代表了多个基因家族,包括表观遗传调节因子、细胞周期相关基因、DNA损伤反应/修复基因、代谢调节因子以及与Wnt、Notch、Hedgehog、JAK/STAT、NF-κB和MAPK信号通路相关的基因。在这42个假定驱动基因中,有已知的驱动基因,如EGFR、ERBB2和PIK3CA。野生型KRAS在几种癌症类型中发生扩增,并且KRAS扩增的癌细胞系对KRAS shRNA最为敏感,这表明KRAS扩增是一个独立的致癌事件。一些MAP激酶衔接蛋白与其受体酪氨酸激酶共同扩增,如FGFR衔接蛋白FRS2以及EGFR家族衔接蛋白GRB2和GRB7。泛素样连接酶DCUN1D1和组蛋白甲基转移酶NSD3也被鉴定为新的假定癌症驱动基因。我们讨论了现有癌症药物靶点的患者定制意义,并进一步探讨了药物研发工作的潜在新机遇。