Li Mei, Xia Zhigui, Yan He
National Institute of Parasitic Diseases, Chinese Centre for Diseases Control and Prevention, Key Laboratory of Parasite and Vector Biology, Ministry of Public Health, WHO Collaborating Centre for Malaria, Schistosomiasis and Filariasis, Shanghai 200025, People's Republic of China.
Malar J. 2014 Jun 3;13:216. doi: 10.1186/1475-2875-13-216.
Plasmodium ovale is relatively unfamiliar to Chinese staff engaged in malaria diagnosis. In 2013, dried blood spots of four unidentified but suspected ovale malaria samples were sent to the National Malaria Reference Laboratory (NMRL) for reconfirmation.
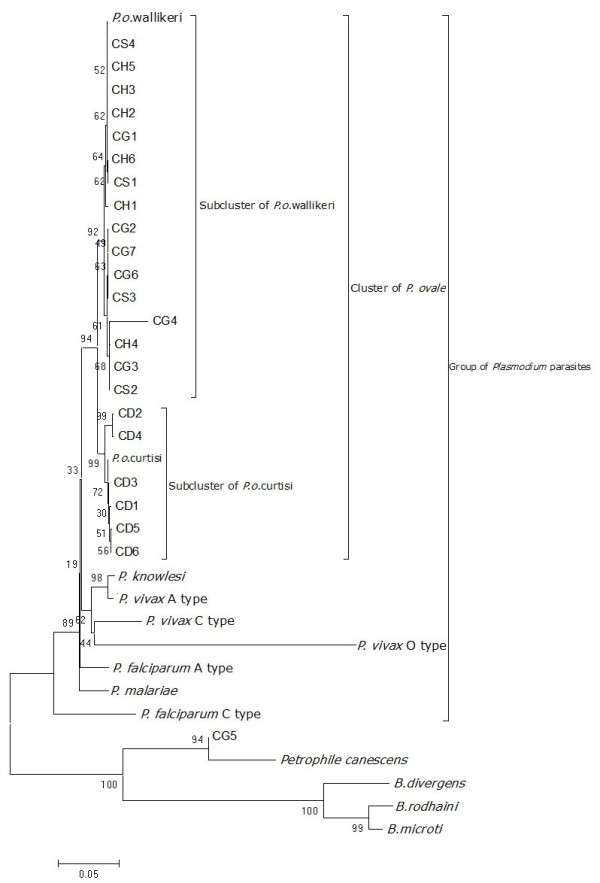
Partial and complete, small, subunit ribosomal DNA (SSU rDNA) sequences of four samples were obtained with PCR-cloning-sequencing method. Obtained sequences were analyzed by aligning with each other and with nine SSU rDNA sequences of six known Plasmodium parasites. A phylogenetic tree was constructed based on complete SSU rDNA sequences and 12 same gene sequences derived from six known Plasmodium parasites and three Babesia parasites. Primary structure of conservative and variable regions of variant sequences was determined also by comparing them with those of six known Plasmodium parasites. To confirm their existence in genome, they were redetected with primers matching their variable regions. PCR systems aimed to roughly detect any eukaryotes and prokaryotes respectively were also applied to search for other pathogens in one of four patients.
Totally, 19 partial and 23 complete SSU rDNA sequences obtained from four samples. Except eight variant sequences, similarities among sequences from same DNA sample were in general high (more than 98%). The phylogenetic analysis revealed that three cases were infected by P. ovale wallikeri and one by P. ovale curtisi. Four of the variant sequences which obtained from four samples relatively showed high similarities with each other (98.5%-100%). Identical variant sequences actually could be re-obtained from each DNA sample. Their primary structure of conservative and variable regions showed quite fit with that of six known Plasmodium parasites. The test for prokaryote pathogens showed negative and the tests for eukaryotes only found DNA sequences of Human and P. ovale parasites.
Both P. ovale wallikeri and P. ovale curtisi infections are present in imported malaria cases of China. New type of partial SSU rDNA sequence which assumed to express in a certain life stage of P. ovale was obtained from both P. ovale wallikeri and P. ovale curtisi samples. This discovery would supply information and clues to identify and understand P. ovale parasites more accurately.
卵形疟原虫对于从事疟疾诊断的中国工作人员来说相对陌生。2013年,四份身份不明但疑似卵形疟的样本干血斑被送至国家疟疾参考实验室(NMRL)进行重新确认。
采用PCR-克隆-测序方法获得四个样本的部分及完整的小亚基核糖体DNA(SSU rDNA)序列。将获得的序列相互比对,并与六种已知疟原虫的九个SSU rDNA序列进行比对分析。基于完整的SSU rDNA序列以及来自六种已知疟原虫和三种巴贝斯虫的12个相同基因序列构建系统发育树。通过将变异序列的保守区和可变区的一级结构与六种已知疟原虫的相应结构进行比较,确定其一级结构。为确认它们在基因组中的存在,使用与可变区匹配的引物对其进行重新检测。还应用分别旨在大致检测任何真核生物和原核生物的PCR系统,在四名患者中的一名患者中寻找其他病原体。
从四个样本中共获得19个部分SSU rDNA序列和23个完整SSU rDNA序列。除了八个变异序列外,来自同一DNA样本的序列之间的相似性总体较高(超过98%)。系统发育分析表明,三例感染的是沃氏卵形疟原虫,一例感染的是柯氏卵形疟原虫。从四个样本中获得的四个变异序列彼此之间相对显示出较高的相似性(98.5%-100%)。实际上可以从每个DNA样本中重新获得相同的变异序列。它们保守区和可变区的一级结构与六种已知疟原虫的结构相当吻合。原核生物病原体检测呈阴性,真核生物检测仅发现人类和卵形疟原虫的DNA序列。
中国输入性疟疾病例中存在沃氏卵形疟原虫和柯氏卵形疟原虫感染。从沃氏卵形疟原虫和柯氏卵形疟原虫样本中均获得了假定在卵形疟原虫某一生命阶段表达的新型部分SSU rDNA序列。这一发现将为更准确地鉴定和了解卵形疟原虫提供信息和线索。