Department of Genome Sciences, University of Washington, Seattle, Washington 98195, USA.
Genome Res. 2012 Sep;22(9):1689-97. doi: 10.1101/gr.134890.111.
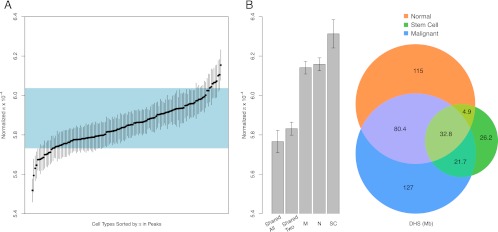
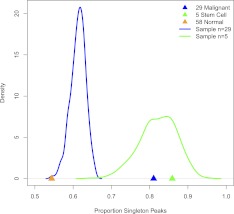
The characteristics and evolutionary forces acting on regulatory variation in humans remains elusive because of the difficulty in defining functionally important noncoding DNA. Here, we combine genome-scale maps of regulatory DNA marked by DNase I hypersensitive sites (DHSs) from 138 cell and tissue types with whole-genome sequences of 53 geographically diverse individuals in order to better delimit the patterns of regulatory variation in humans. We estimate that individuals likely harbor many more functionally important variants in regulatory DNA compared with protein-coding regions, although they are likely to have, on average, smaller effect sizes. Moreover, we demonstrate that there is significant heterogeneity in the level of functional constraint in regulatory DNA among different cell types. We also find marked variability in functional constraint among transcription factor motifs in regulatory DNA, with sequence motifs for major developmental regulators, such as HOX proteins, exhibiting levels of constraint comparable to protein-coding regions. Finally, we perform a genome-wide scan of recent positive selection and identify hundreds of novel substrates of adaptive regulatory evolution that are enriched for biologically interesting pathways such as melanogenesis and adipocytokine signaling. These data and results provide new insights into patterns of regulatory variation in individuals and populations and demonstrate that a large proportion of functionally important variation lies beyond the exome.
由于难以定义功能重要的非编码 DNA,人类调控变异的特征和进化力量仍然难以捉摸。在这里,我们将来自 138 种细胞和组织类型的标记有 DNase I 超敏位点 (DHS) 的调控 DNA 的基因组规模图谱与 53 个具有地理多样性的个体的全基因组序列相结合,以便更好地划定人类调控变异的模式。我们估计,与蛋白质编码区域相比,个体可能在调控 DNA 中拥有更多的功能重要变体,尽管它们的平均效应大小可能较小。此外,我们证明在不同细胞类型中,调控 DNA 的功能约束水平存在显著的异质性。我们还发现,调控 DNA 中转录因子基序的功能约束存在明显的可变性,主要发育调节剂(如 HOX 蛋白)的序列基序表现出与蛋白质编码区域相当的约束水平。最后,我们对近期正选择进行了全基因组扫描,鉴定出数百个新的适应性调控进化的底物,这些底物富集了生物有趣的途径,如黑色素生成和脂肪细胞因子信号转导。这些数据和结果为个体和群体的调控变异模式提供了新的见解,并表明很大一部分功能重要的变异位于外显子之外。