Mose Lisle E, Wilkerson Matthew D, Hayes D Neil, Perou Charles M, Parker Joel S
Lineberger Comprehensive Cancer Center, Department of Genetics, Division of Medical Oncology, Department of Internal Medicine, Multidisciplinary Thoracic Oncology Program, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Lineberger Comprehensive Cancer Center, Department of Genetics, Division of Medical Oncology, Department of Internal Medicine, Multidisciplinary Thoracic Oncology Program, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA Lineberger Comprehensive Cancer Center, Department of Genetics, Division of Medical Oncology, Department of Internal Medicine, Multidisciplinary Thoracic Oncology Program, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Bioinformatics. 2014 Oct;30(19):2813-5. doi: 10.1093/bioinformatics/btu376. Epub 2014 Jun 6.
Variant detection from next-generation sequencing (NGS) data is an increasingly vital aspect of disease diagnosis, treatment and research. Commonly used NGS-variant analysis tools generally rely on accurately mapped short reads to identify somatic variants and germ-line genotypes. Existing NGS read mappers have difficulty accurately mapping short reads containing complex variation (i.e. more than a single base change), thus making identification of such variants difficult or impossible. Insertions and deletions (indels) in particular have been an area of great difficulty. Indels are frequent and can have substantial impact on function, which makes their detection all the more imperative.
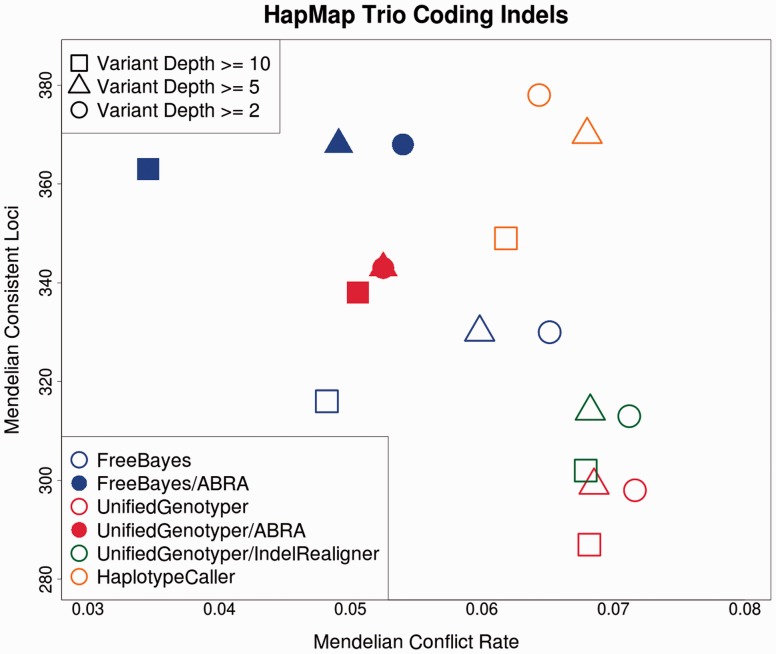
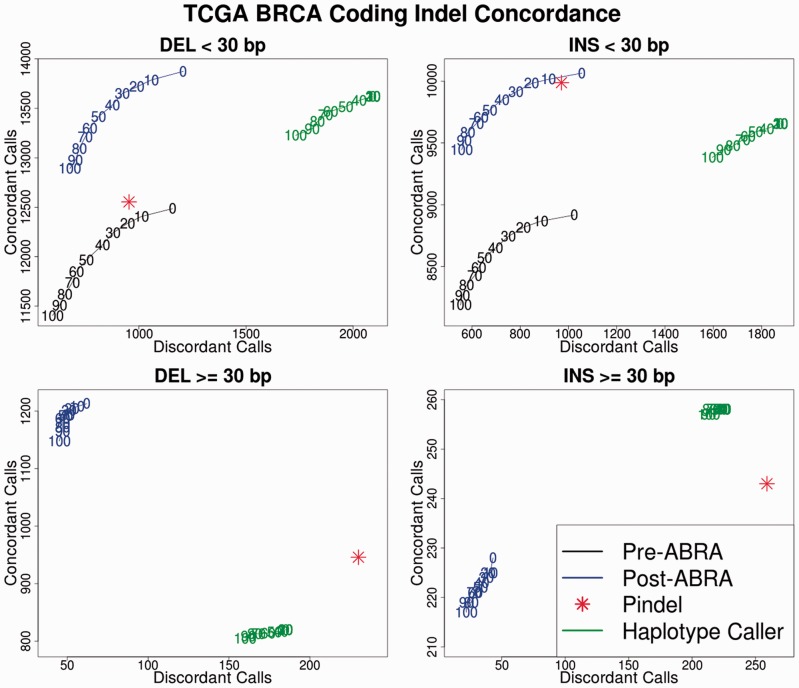
We present ABRA, an assembly-based realigner, which uses an efficient and flexible localized de novo assembly followed by global realignment to more accurately remap reads. This results in enhanced performance for indel detection as well as improved accuracy in variant allele frequency estimation.
ABRA is implemented in a combination of Java and C/C++ and is freely available for download at https://github.com/mozack/abra.
从下一代测序(NGS)数据中检测变异是疾病诊断、治疗和研究中日益重要的一个方面。常用的NGS变异分析工具通常依赖于精确映射的短读段来识别体细胞变异和种系基因型。现有的NGS读段映射器难以精确映射包含复杂变异(即不止一个碱基变化)的短读段,因此使得此类变异的识别变得困难或不可能。特别是插入和缺失(indel)一直是一个极具难度的领域。Indel很常见,并且会对功能产生重大影响,这使得它们的检测更加迫切。
我们提出了ABRA,一种基于组装的重比对器,它使用高效且灵活的局部从头组装,然后进行全局重比对,以更准确地重新映射读段。这导致indel检测性能增强,以及变异等位基因频率估计的准确性提高。
ABRA用Java和C/C++组合实现,可在https://github.com/mozack/abra上免费下载。