Chen X, Paranjape T, Stahlhut C, McVeigh T, Keane F, Nallur S, Miller N, Kerin M, Deng Y, Yao X, Zhao H, Weidhaas J B, Slack F J
1] Department of Molecular, Cellular and Developmental Biology, Yale University, New Haven, CT, USA [2] Program in Computational Biology and Bioinformatics, Yale University School of Medicine, New Haven, CT, USA.
Department of Therapeutic Radiology, Yale University School of Medicine, New Haven, CT, USA.
Oncogene. 2015 Apr 16;34(16):2125-37. doi: 10.1038/onc.2014.117. Epub 2014 Jun 9.
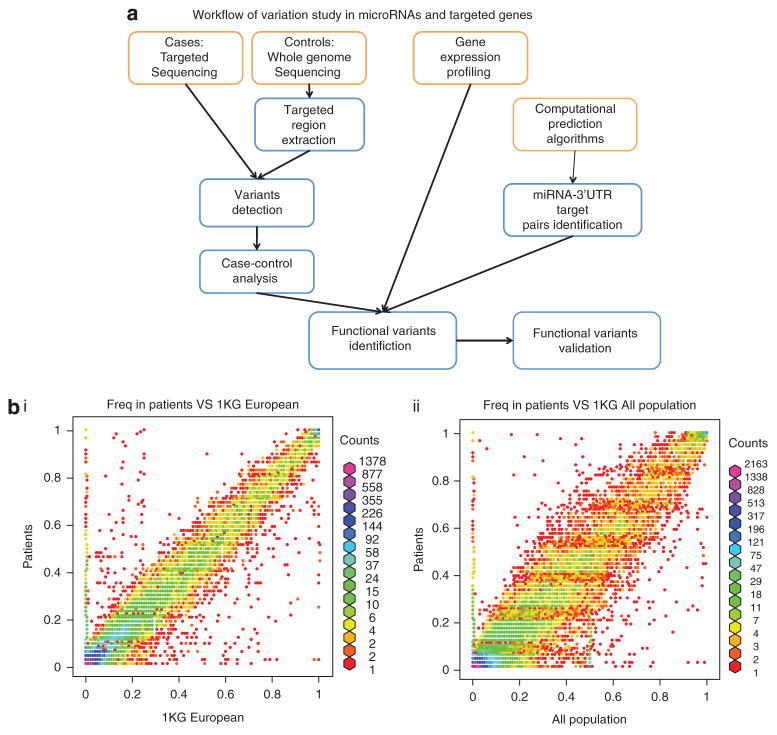
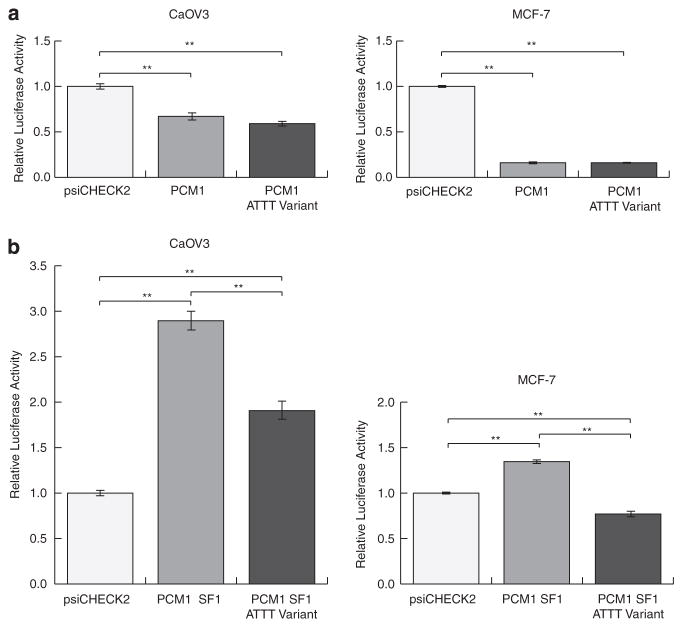
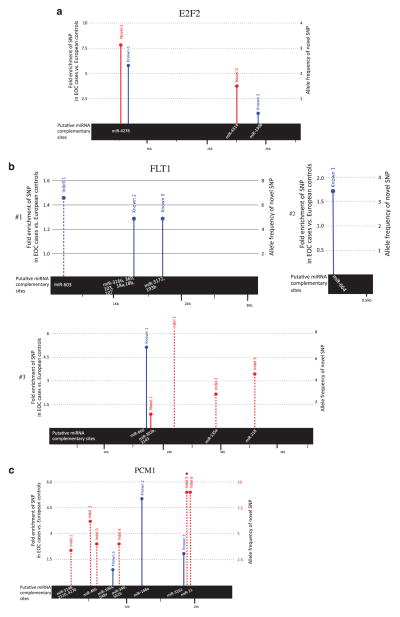
Ovarian cancer is a major cause of cancer deaths, yet there have been few known genetic risk factors identified, the best known of which are disruptions in protein coding sequences (BRCA1 and 2). Recent findings indicate that there are powerful genetic markers of cancer risk outside of these regions, in the noncoding mRNA control regions. To identify additional cancer-associated, functional non-protein-coding sequence germline variants associated with ovarian cancer risk, we captured DNA regions corresponding to all validated human microRNAs and the 3' untranslated regions (UTRs) of ~6000 cancer-associated genes from 31 ovarian cancer patients. Multiple single-nucleotide polymorphisms in the 3'UTR of the vascular endothelial growth factor receptor/FLT1, E2F2 and PCM1 oncogenes were highly enriched in ovarian cancer patients compared with the 1000 Genome Project. Sequenom validation in a case-control study (267 cases and 89 controls) confirmed a novel variant in the PCM1 3'UTR is significantly associated with ovarian cancer (P=0.0086). This work identifies a potential new ovarian cancer locus and further confirms that cancer resequencing efforts should not ignore the study of noncoding regions of cancer patients.
卵巢癌是癌症死亡的主要原因之一,但已知的遗传风险因素却很少,其中最广为人知的是蛋白质编码序列(BRCA1和2)的破坏。最近的研究结果表明,在这些区域之外的非编码mRNA控制区域存在强大的癌症风险遗传标记。为了识别与卵巢癌风险相关的其他癌症相关功能性非蛋白质编码序列种系变体,我们从31名卵巢癌患者中捕获了与所有已验证的人类微小RNA以及约6000个癌症相关基因的3'非翻译区(UTR)相对应的DNA区域。与千人基因组计划相比,血管内皮生长因子受体/FLT1、E2F2和PCM1癌基因3'UTR中的多个单核苷酸多态性在卵巢癌患者中高度富集。在一项病例对照研究(267例病例和89例对照)中的Sequenom验证证实,PCM1 3'UTR中的一个新变体与卵巢癌显著相关(P=0.0086)。这项工作确定了一个潜在的新卵巢癌基因座,并进一步证实癌症重测序研究不应忽视对癌症患者非编码区域的研究。