Huang Shunping, Holt James, Kao Chia-Yu, McMillan Leonard, Wang Wei
Department of Computer Science, University of North Carolina, Chapel Hill, NC 27599, Department of Computer Science, University of California, Los Angeles, CA 90095, USA.
Department of Computer Science, University of North Carolina, Chapel Hill, NC 27599, Department of Computer Science, University of California, Los Angeles, CA 90095, USA
Database (Oxford). 2014 Jun 18;2014. doi: 10.1093/database/bau057. Print 2014.
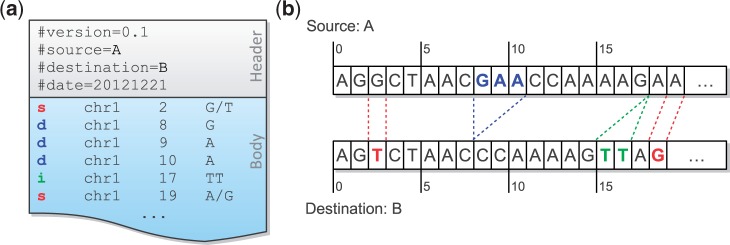
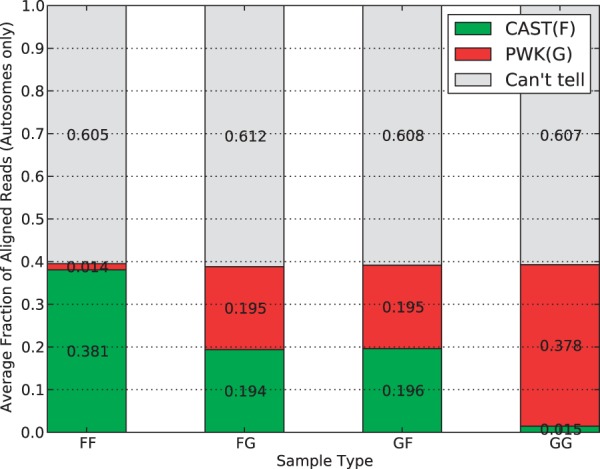
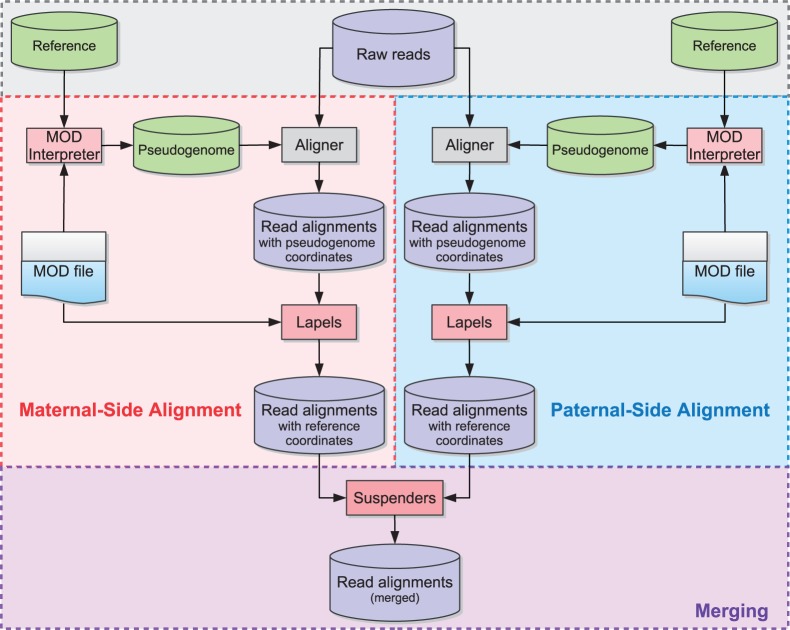
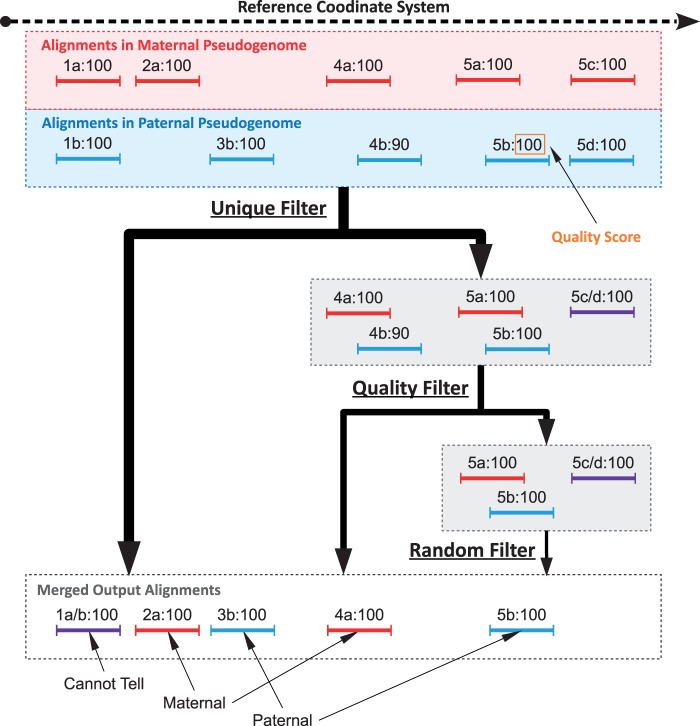
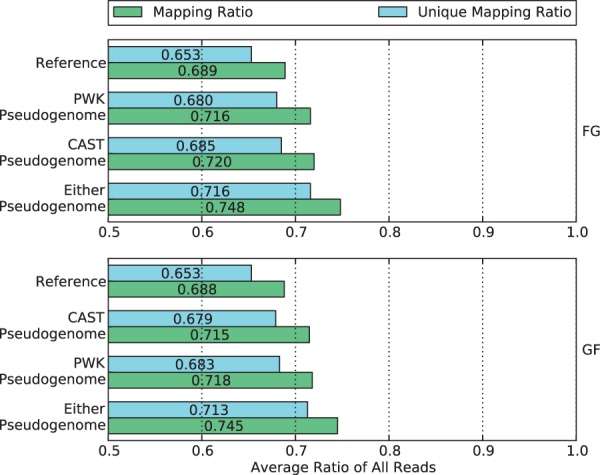
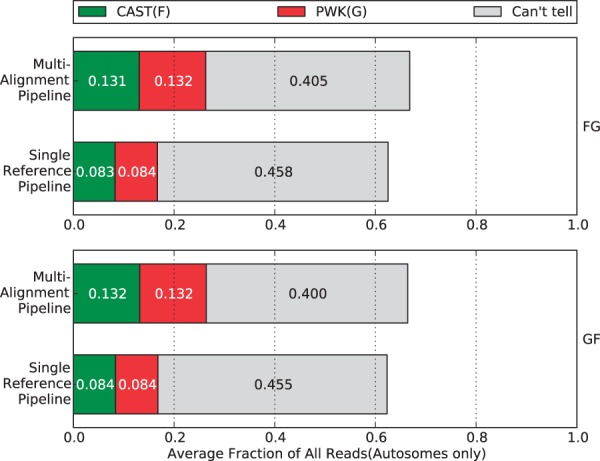
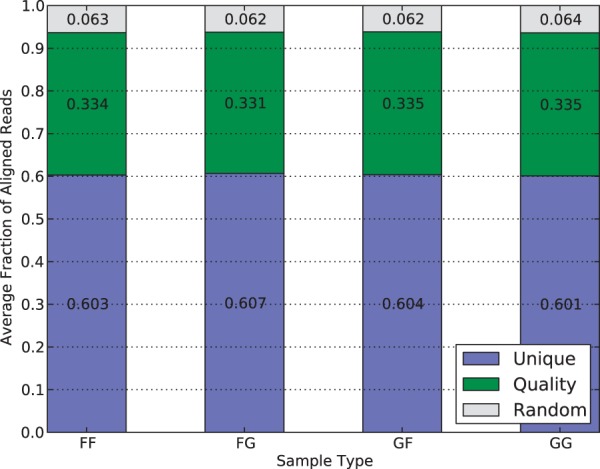
Mapping reads to a reference sequence is a common step when analyzing allele effects in high-throughput sequencing data. The choice of reference is critical because its effect on quantitative sequence analysis is non-negligible. Recent studies suggest aligning to a single standard reference sequence, as is common practice, can lead to an underlying bias depending on the genetic distances of the target sequences from the reference. To avoid this bias, researchers have resorted to using modified reference sequences. Even with this improvement, various limitations and problems remain unsolved, which include reduced mapping ratios, shifts in read mappings and the selection of which variants to include to remove biases. To address these issues, we propose a novel and generic multi-alignment pipeline. Our pipeline integrates the genomic variations from known or suspected founders into separate reference sequences and performs alignments to each one. By mapping reads to multiple reference sequences and merging them afterward, we are able to rescue more reads and diminish the bias caused by using a single common reference. Moreover, the genomic origin of each read is determined and annotated during the merging process, providing a better source of information to assess differential expression than simple allele queries at known variant positions. Using RNA-seq of a diallel cross, we compare our pipeline with the single-reference pipeline and demonstrate our advantages of more aligned reads and a higher percentage of reads with assigned origins. Database URL: http://csbio.unc.edu/CCstatus/index.py?run=Pseudo.
在分析高通量测序数据中的等位基因效应时,将 reads 映射到参考序列是一个常见步骤。参考序列的选择至关重要,因为其对定量序列分析的影响不可忽视。最近的研究表明,按照常规做法与单个标准参考序列进行比对,可能会导致潜在偏差,这取决于目标序列与参考序列的遗传距离。为避免这种偏差,研究人员已采用修改后的参考序列。即便有了这一改进,各种局限性和问题仍未解决,其中包括映射率降低、读取映射偏移以及选择要纳入哪些变体以消除偏差。为解决这些问题,我们提出了一种新颖且通用的多比对流程。我们的流程将来自已知或疑似奠基者的基因组变异整合到单独的参考序列中,并对每个序列进行比对。通过将 reads 映射到多个参考序列并随后合并它们,我们能够挽救更多 reads,并减少因使用单个通用参考序列而导致的偏差。此外,在合并过程中确定并注释每个 read 的基因组来源,这比在已知变异位置进行简单的等位基因查询能提供更好的信息来源以评估差异表达。使用双列杂交的 RNA-seq,我们将我们的流程与单参考序列流程进行比较,并展示了我们在更多比对 reads 和更高比例有指定来源的 reads 方面的优势。数据库网址:http://csbio.unc.edu/CCstatus/index.py?run=Pseudo