Department of Botany and Plant Pathology, Oregon State University, Corvallis, Oregon, United States of America.
PLoS One. 2011;6(10):e25279. doi: 10.1371/journal.pone.0025279. Epub 2011 Oct 6.
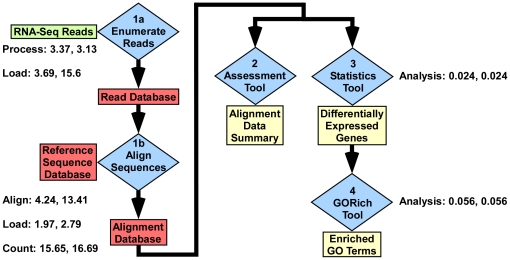
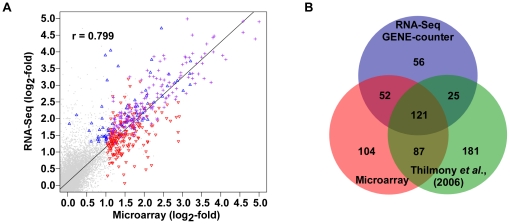
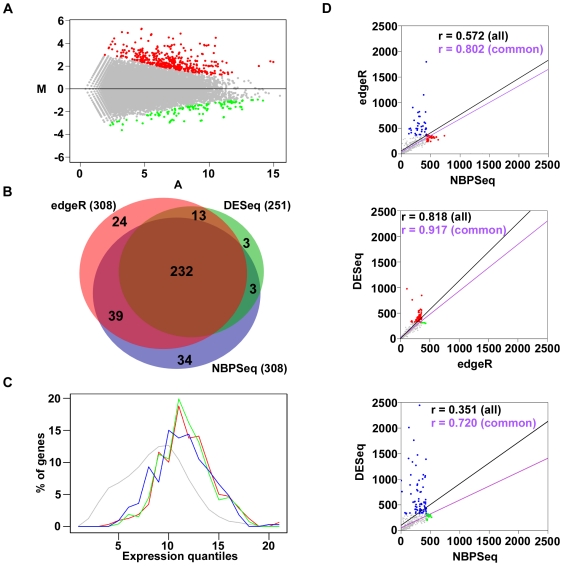
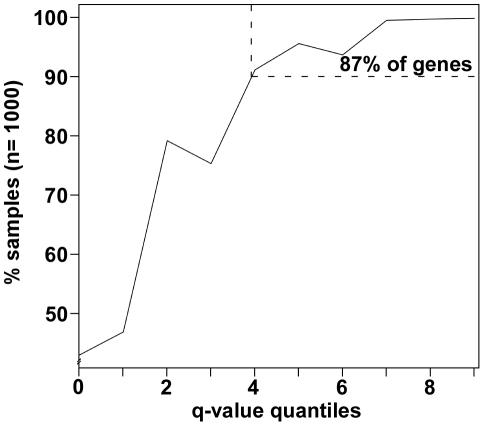
GENE-counter is a complete Perl-based computational pipeline for analyzing RNA-Sequencing (RNA-Seq) data for differential gene expression. In addition to its use in studying transcriptomes of eukaryotic model organisms, GENE-counter is applicable for prokaryotes and non-model organisms without an available genome reference sequence. For alignments, GENE-counter is configured for CASHX, Bowtie, and BWA, but an end user can use any Sequence Alignment/Map (SAM)-compliant program of preference. To analyze data for differential gene expression, GENE-counter can be run with any one of three statistics packages that are based on variations of the negative binomial distribution. The default method is a new and simple statistical test we developed based on an over-parameterized version of the negative binomial distribution. GENE-counter also includes three different methods for assessing differentially expressed features for enriched gene ontology (GO) terms. Results are transparent and data are systematically stored in a MySQL relational database to facilitate additional analyses as well as quality assessment. We used next generation sequencing to generate a small-scale RNA-Seq dataset derived from the heavily studied defense response of Arabidopsis thaliana and used GENE-counter to process the data. Collectively, the support from analysis of microarrays as well as the observed and substantial overlap in results from each of the three statistics packages demonstrates that GENE-counter is well suited for handling the unique characteristics of small sample sizes and high variability in gene counts.
GENE-counter 是一个完整的基于 Perl 的计算流程,用于分析 RNA-Seq(RNA 测序)数据以进行差异基因表达分析。除了在真核模式生物的转录组研究中使用外,GENE-counter 还适用于没有可用基因组参考序列的原核生物和非模式生物。对于比对,GENE-counter 配置为 CASHX、Bowtie 和 BWA,但用户可以使用任何首选的符合序列比对/映射 (SAM) 的程序。为了分析差异基因表达数据,GENE-counter 可以与基于负二项式分布变体的三个统计软件包中的任何一个一起运行。默认方法是我们基于负二项式分布的过参数化版本开发的新的简单统计测试。GENE-counter 还包括三种用于评估富含基因本体论 (GO) 术语的差异表达特征的方法。结果是透明的,数据系统地存储在 MySQL 关系数据库中,以方便进行额外的分析和质量评估。我们使用下一代测序生成了一个源自拟南芥大量研究的防御反应的小规模 RNA-Seq 数据集,并使用 GENE-counter 处理数据。总的来说,微阵列分析的支持以及来自三个统计软件包中的每一个的观察到的和实质性的重叠结果表明,GENE-counter 非常适合处理小样本量和基因计数高度变化的独特特征。