Ocholla Harold, Preston Mark D, Mipando Mwapatsa, Jensen Anja T R, Campino Susana, MacInnis Bronwyn, Alcock Daniel, Terlouw Anja, Zongo Issaka, Oudraogo Jean-Bosco, Djimde Abdoulaye A, Assefa Samuel, Doumbo Ogobara K, Borrmann Steffen, Nzila Alexis, Marsh Kevin, Fairhurst Rick M, Nosten Francois, Anderson Tim J C, Kwiatkowski Dominic P, Craig Alister, Clark Taane G, Montgomery Jacqui
Malawi-Liverpool-Wellcome Trust Clinical Research Programme Liverpool School of Tropical Medicine, Pembroke Place, Liverpool.
Faculty of Infectious and Tropical Diseases, London School of Hygiene and Tropical Medicine.
J Infect Dis. 2014 Dec 15;210(12):1991-2000. doi: 10.1093/infdis/jiu349. Epub 2014 Jun 19.
Selection by host immunity and antimalarial drugs has driven extensive adaptive evolution in Plasmodium falciparum and continues to produce ever-changing landscapes of genetic variation.
We performed whole-genome sequencing of 69 P. falciparum isolates from Malawi and used population genetics approaches to investigate genetic diversity and population structure and identify loci under selection.
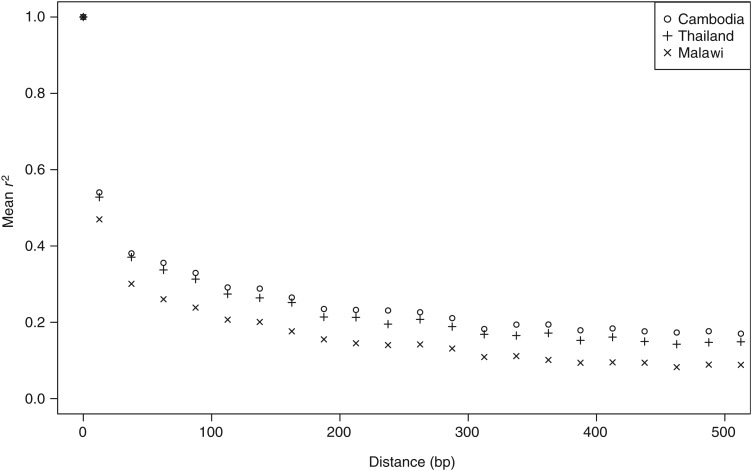
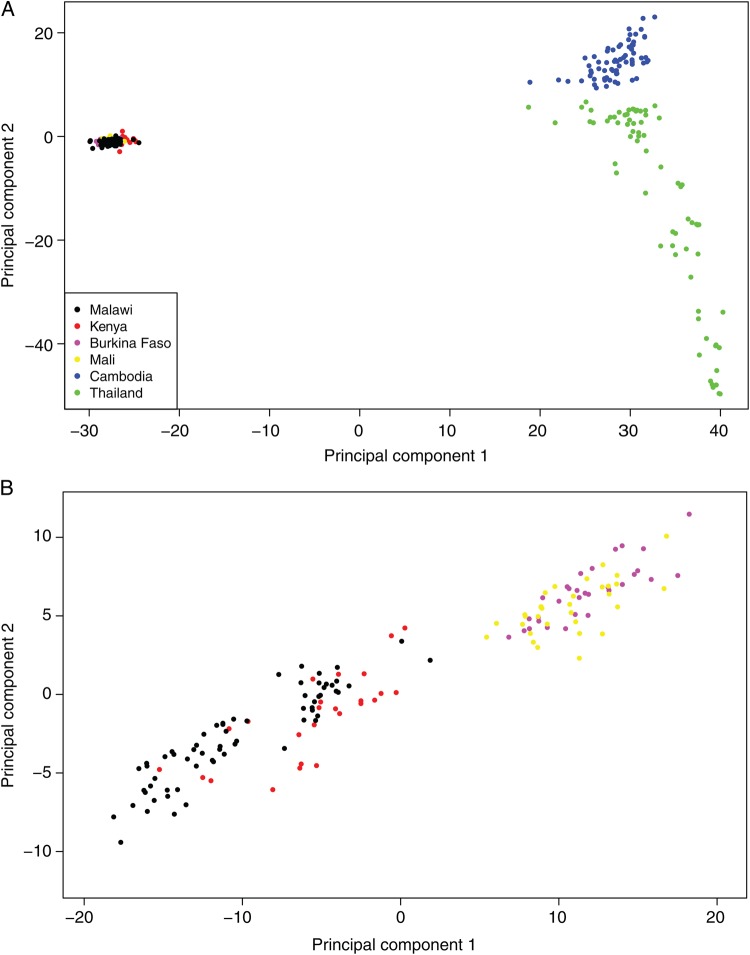
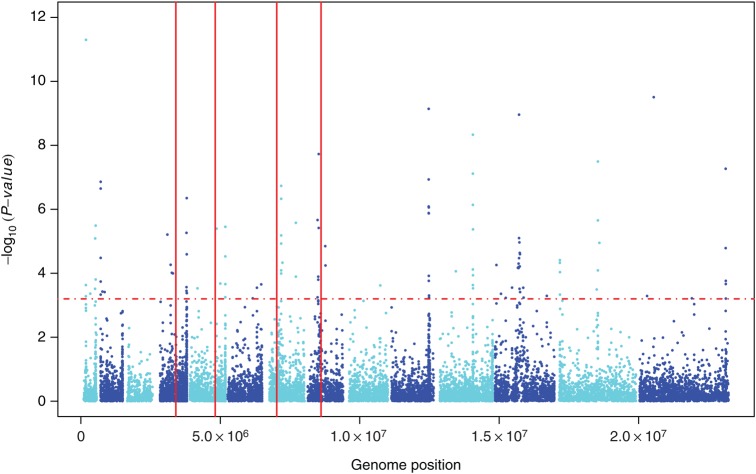
High genetic diversity (π = 2.4 × 10(-4)), moderately high multiplicity of infection (2.7), and low linkage disequilibrium (500-bp) were observed in Chikhwawa District, Malawi, an area of high malaria transmission. Allele frequency-based tests provided evidence of recent population growth in Malawi and detected potential targets of host immunity and candidate vaccine antigens. Comparison of the sequence variation between isolates from Malawi and those from 5 geographically dispersed countries (Kenya, Burkina Faso, Mali, Cambodia, and Thailand) detected population genetic differences between Africa and Asia, within Southeast Asia, and within Africa. Haplotype-based tests of selection to sequence data from all 6 populations identified signals of directional selection at known drug-resistance loci, including pfcrt, pfdhps, pfmdr1, and pfgch1.
The sequence variations observed at drug-resistance loci reflect differences in each country's historical use of antimalarial drugs and may be useful in formulating local malaria treatment guidelines.
宿主免疫和抗疟药物的选择推动了恶性疟原虫广泛的适应性进化,并持续产生不断变化的遗传变异格局。
我们对来自马拉维的69株恶性疟原虫分离株进行了全基因组测序,并使用群体遗传学方法研究遗传多样性和群体结构,以及识别受选择的基因座。
在疟疾传播率高的马拉维奇夸瓦区,观察到高遗传多样性(π = 2.4 × 10(-4))、中等偏高的感染复数(2.7)和低连锁不平衡(500bp)。基于等位基因频率的测试为马拉维近期的群体增长提供了证据,并检测到宿主免疫的潜在靶点和候选疫苗抗原。比较来自马拉维的分离株与来自5个地理上分散的国家(肯尼亚、布基纳法索、马里、柬埔寨和泰国)的分离株之间的序列变异,发现了非洲和亚洲之间、东南亚内部以及非洲内部的群体遗传差异。对来自所有6个群体的序列数据进行基于单倍型的选择测试,在已知的耐药基因座,包括pfcrt、pfdhps、pfmdr1和pfgch1,识别出定向选择信号。
在耐药基因座观察到的序列变异反映了每个国家抗疟药物的历史使用差异,可能有助于制定当地的疟疾治疗指南。