Fu Wei, Wang Kai, Zhao Jun-Long, Yu Heng-Chao, Li San-Zhong, Lin Yan, Liang Liang, Huang Si-Yong, Liang Ying-Min, Han Hua, Qin Hong-Yan
Department of Hematology, Tangdu Hospital, Fourth Military Medical University, Xi'an 710038, People's Republic of China.
BMC Cancer. 2014 Jun 22;14:463. doi: 10.1186/1471-2407-14-463.
Aberrantly activated Notch signaling has been found in more than 50% of patients with T-cell acute lymphoblastic leukemia (T-ALL). Current strategies that employ γ-secretase inhibitors (GSIs) to target Notch activation have not been successful. Many limitations, such as non-Notch specificity, dose-limiting gastrointestinal toxicity and GSI resistance, have prompted an urgent need for more effective Notch signaling inhibitors for T-ALL treatment. Human four-and-a-half LIM domain protein 1C (FHL1C) (KyoT2 in mice) has been demonstrated to suppress Notch activation in vitro, suggesting that FHL1C may be new candidate target in T-ALL therapy. However, the role of FHL1C in T-ALL cells remained unclear.
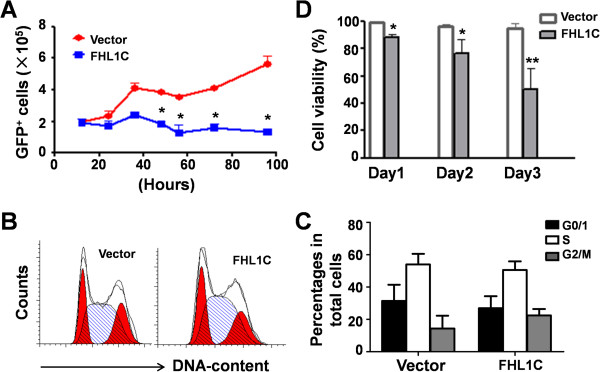
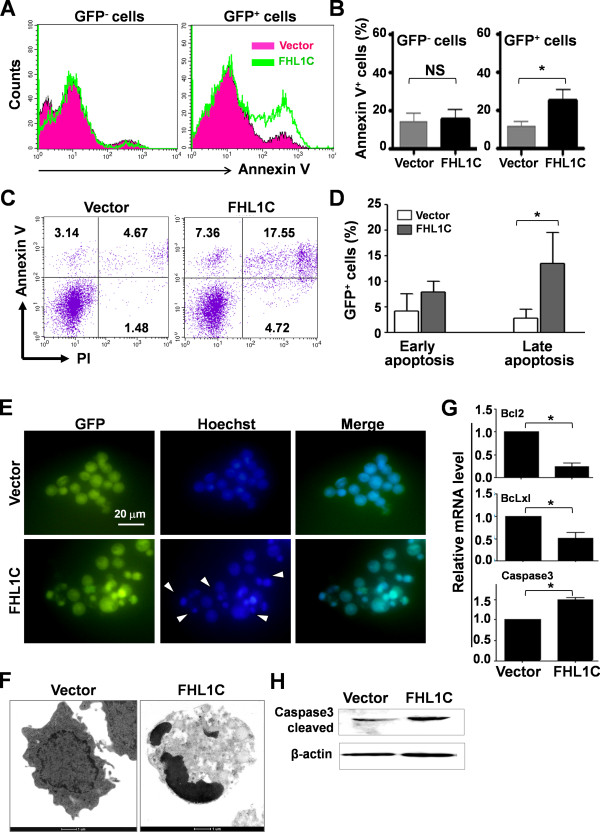
Using RT-PCR, we amplified full-length human FHL1C, and constructed full-length and various truncated forms of FHL1C. Using cell transfection, flow cytometry, transmission electron microscope, real-time RT-PCR, and Western blotting, we found that overexpression of FHL1C induced apoptosis of Jurkat cells. By using a reporter assay and Annexin-V staining, the minimal functional sequence of FHL1C inhibiting RBP-J-mediated Notch transactivation and inducing cell apoptosis was identified. Using real-time PCR and Western blotting, we explored the possible molecular mechanism of FHL1C-induced apoptosis. All data were statistically analyzed with the SPSS version 12.0 software.
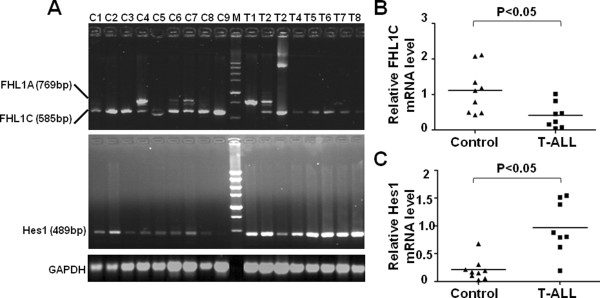
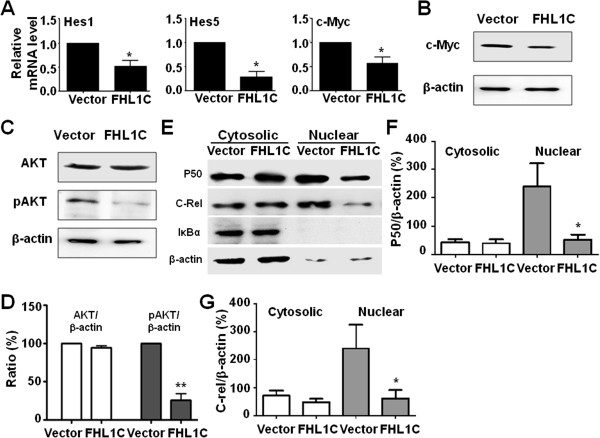
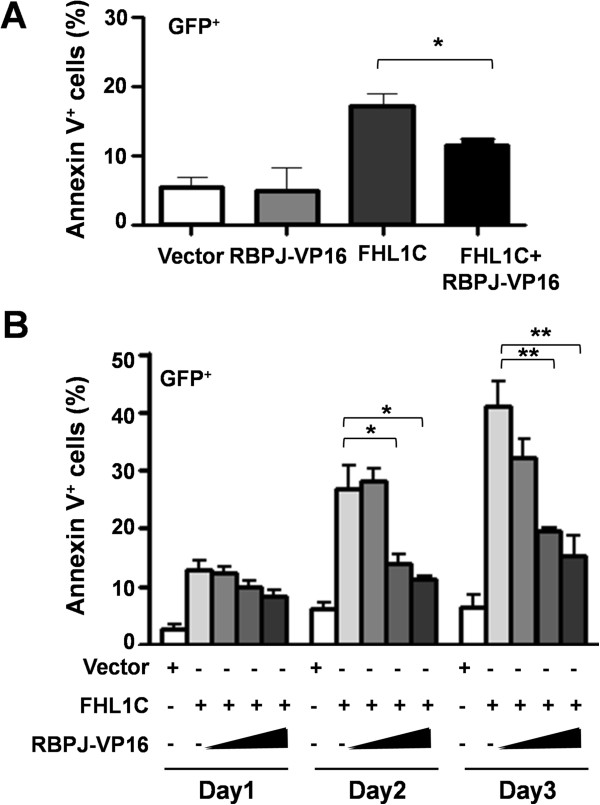
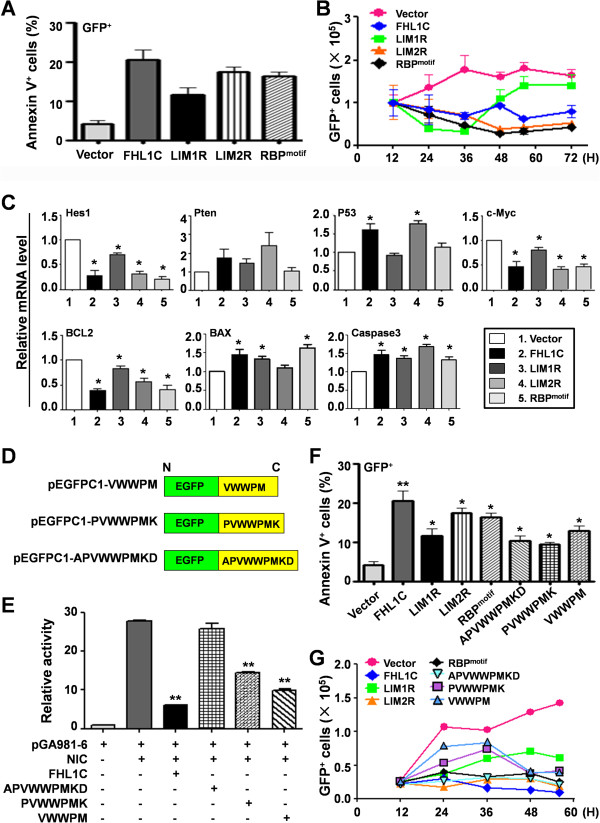
In Jurkat cells derived from a Notch1-associated T-ALL cell line insensitive to GSI treatment, we observed that overexpression of FHL1C, which is down-regulated in T-ALL patients, strongly induced apoptosis. Furthermore, we verified that FHL1C-induced apoptosis depended on the RBP-J-binding motif at the C-terminus of FHL1C. Using various truncated forms of FHL1C, we found that the RBP-J-binding motif of FHL1C had almost the same effect as full-length FHL1C on the induction of apoptosis, suggesting that the minimal functional sequence in the RBP-J-binding motif of FHL1C might be a new drug candidate for T-ALL treatment. We also explored the molecular mechanism of FHL1C overexpression-induced apoptosis, which suppressed downstream target genes such as Hes1 and c-Myc and key signaling pathways such as PI3K/AKT and NF-κB of Notch signaling involved in T-ALL progression.
Our study has revealed that FHL1C overexpression induces Jurkat cell apoptosis. This finding may provide new insights in designing new Notch inhibitors based on FHL1C to treat T-ALL.
在超过50%的T细胞急性淋巴细胞白血病(T-ALL)患者中发现Notch信号异常激活。目前采用γ-分泌酶抑制剂(GSIs)靶向Notch激活的策略尚未成功。许多局限性,如非Notch特异性、剂量限制性胃肠道毒性和GSI耐药性,促使迫切需要更有效的Notch信号抑制剂来治疗T-ALL。人类四又二分之一LIM结构域蛋白1C(FHL1C)(小鼠中的KyoT2)已被证明在体外可抑制Notch激活,这表明FHL1C可能是T-ALL治疗中的新候选靶点。然而,FHL1C在T-ALL细胞中的作用仍不清楚。
使用逆转录聚合酶链反应(RT-PCR)扩增人FHL1C全长,并构建FHL1C的全长及各种截短形式。通过细胞转染、流式细胞术、透射电子显微镜、实时RT-PCR和蛋白质免疫印迹法,我们发现FHL1C过表达可诱导Jurkat细胞凋亡。通过报告基因检测和膜联蛋白V染色,确定了FHL1C抑制RBP-J介导的Notch反式激活并诱导细胞凋亡的最小功能序列。使用实时PCR和蛋白质免疫印迹法,我们探索了FHL1C诱导凋亡的可能分子机制。所有数据均使用SPSS 12.0软件进行统计分析。
在源自对GSI治疗不敏感的Notch1相关T-ALL细胞系的Jurkat细胞中,我们观察到在T-ALL患者中下调的FHL1C过表达强烈诱导凋亡。此外,我们证实FHL1C诱导的凋亡依赖于FHL1C C末端的RBP-J结合基序。使用FHL1C的各种截短形式,我们发现FHL1C的RBP-J结合基序在诱导凋亡方面与全长FHL1C几乎具有相同的效果,这表明FHL1C的RBP-J结合基序中的最小功能序列可能是T-ALL治疗的新候选药物。我们还探索了FHL1C过表达诱导凋亡的分子机制,其抑制了下游靶基因如Hes1和c-Myc以及参与T-ALL进展的Notch信号的关键信号通路如PI3K/AKT和NF-κB。
我们的研究表明FHL1C过表达可诱导Jurkat细胞凋亡。这一发现可能为基于FHL1C设计新的Notch抑制剂治疗T-ALL提供新的见解。