Menalled Liliana B, Kudwa Andrea E, Oakeshott Steve, Farrar Andrew, Paterson Neil, Filippov Igor, Miller Sam, Kwan Mei, Olsen Michael, Beltran Jose, Torello Justin, Fitzpatrick Jon, Mushlin Richard, Cox Kimberly, McConnell Kristi, Mazzella Matthew, He Dansha, Osborne Georgina F, Al-Nackkash Rand, Bates Gill P, Tuunanen Pasi, Lehtimaki Kimmo, Brunner Dani, Ghavami Afshin, Ramboz Sylvie, Park Larry, Macdonald Douglas, Munoz-Sanjuan Ignacio, Howland David
PsychoGenics Inc., Tarrytown, New York, United States of America.
Department of Medical and Molecular Genetics, King's College London, London, United Kingdom.
PLoS One. 2014 Jun 23;9(6):e99520. doi: 10.1371/journal.pone.0099520. eCollection 2014.
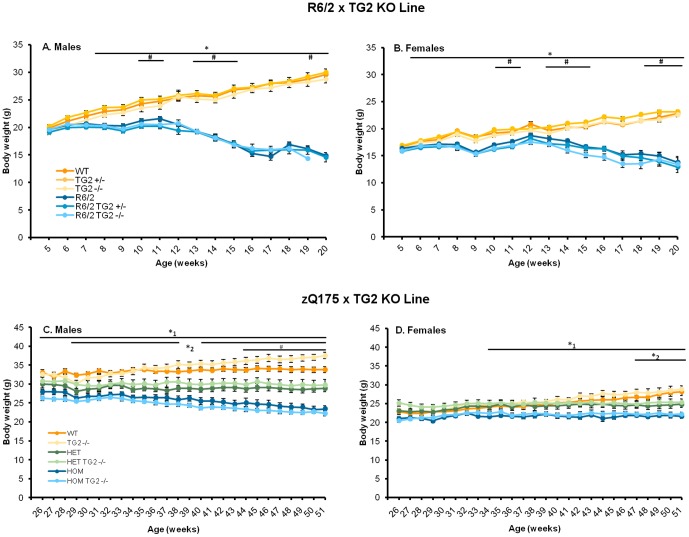
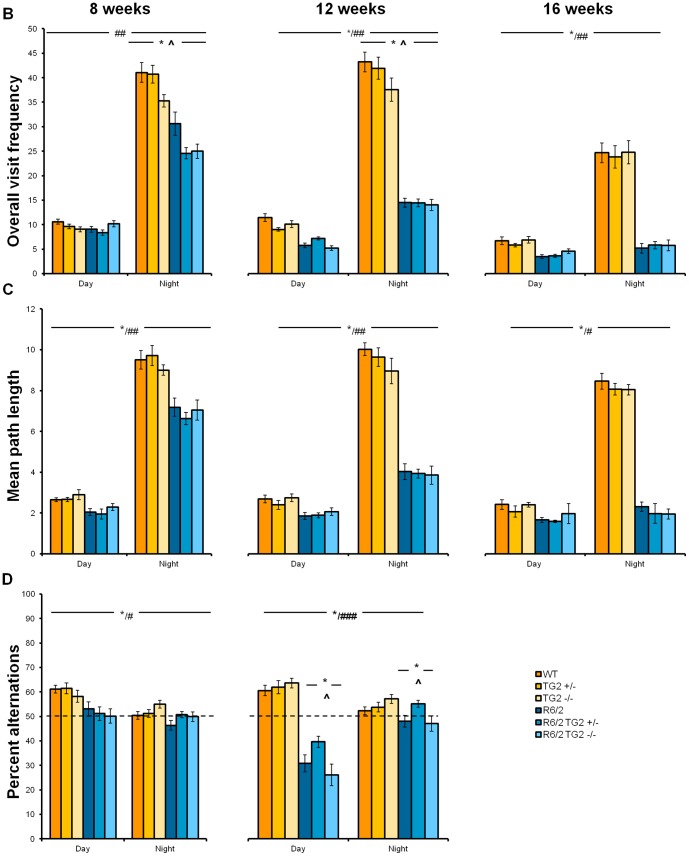
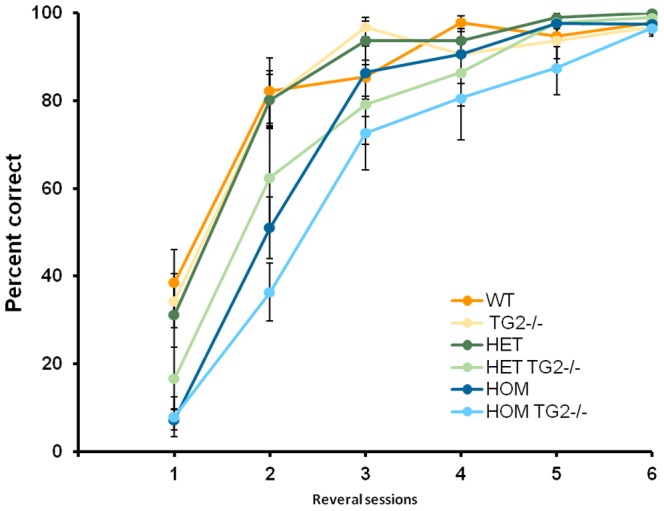
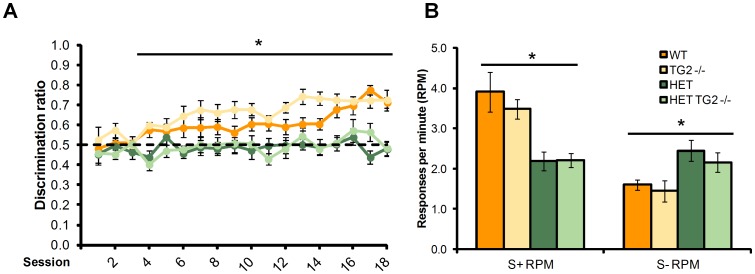
Huntington's disease (HD) is an autosomal dominant, progressive neurodegenerative disorder caused by expansion of CAG repeats in the huntingtin gene. Tissue transglutaminase 2 (TG2), a multi-functional enzyme, was found to be increased both in HD patients and in mouse models of the disease. Furthermore, beneficial effects have been reported from the genetic ablation of TG2 in R6/2 and R6/1 mouse lines. To further evaluate the validity of this target for the treatment of HD, we examined the effects of TG2 deletion in two genetic mouse models of HD: R6/2 CAG 240 and zQ175 knock in (KI). Contrary to previous reports, under rigorous experimental conditions we found that TG2 ablation had no effect on either motor or cognitive deficits, or on the weight loss. In addition, under optimal husbandry conditions, TG2 ablation did not extend R6/2 lifespan. Moreover, TG2 deletion did not change the huntingtin aggregate load in cortex or striatum and did not decrease the brain atrophy observed in either mouse line. Finally, no amelioration of the dysregulation of striatal and cortical gene markers was detected. We conclude that TG2 is not a valid therapeutic target for the treatment of HD.
亨廷顿舞蹈症(HD)是一种常染色体显性遗传的进行性神经退行性疾病,由亨廷顿基因中CAG重复序列的扩增引起。组织转谷氨酰胺酶2(TG2)是一种多功能酶,在HD患者和该疾病的小鼠模型中均被发现有所增加。此外,据报道,在R6/2和R6/1小鼠品系中对TG2进行基因敲除具有有益效果。为了进一步评估该靶点用于治疗HD的有效性,我们在两种HD基因小鼠模型中研究了TG2缺失的影响:R6/2 CAG 240和zQ175基因敲入(KI)模型。与之前的报道相反,在严格的实验条件下,我们发现TG2基因敲除对运动或认知缺陷以及体重减轻均无影响。此外,在最佳饲养条件下,TG2基因敲除并未延长R6/2小鼠的寿命。而且,TG2缺失并未改变皮质或纹状体中亨廷顿蛋白聚集体的负荷,也未减轻在任一小鼠品系中观察到的脑萎缩。最后,未检测到纹状体和皮质基因标志物失调的改善情况。我们得出结论,TG2不是治疗HD的有效治疗靶点。