King's College London, Institute of Psychiatry, Department of Neuroscience, London, United Kingdom ; King's College London, Department of Medical and Molecular Genetics, London, United Kingdom.
King's College London, Department of Neuroimaging, Institute of Psychiatry, London, United Kingdom.
PLoS One. 2013 Dec 19;8(12):e84726. doi: 10.1371/journal.pone.0084726. eCollection 2013.
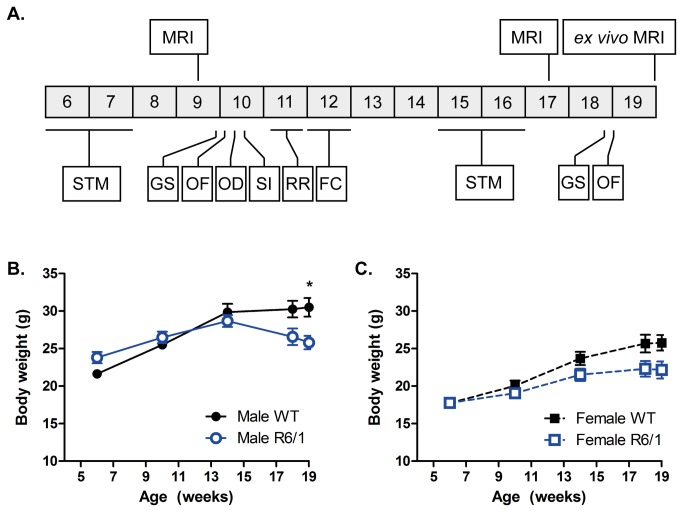
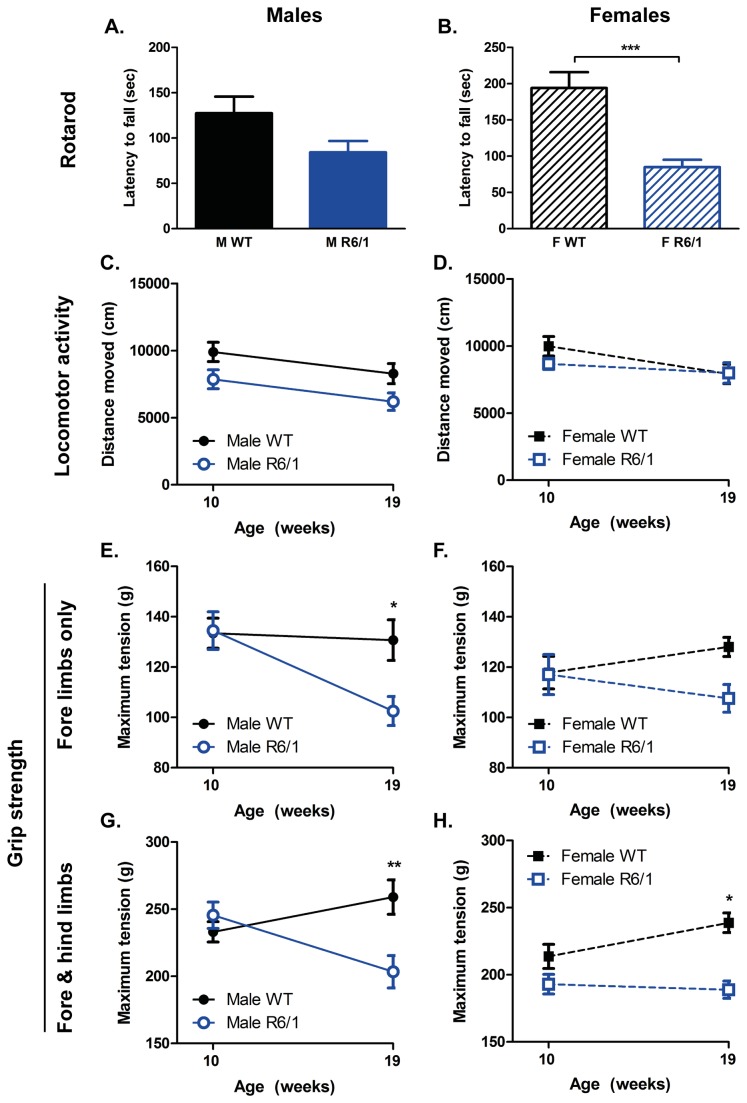
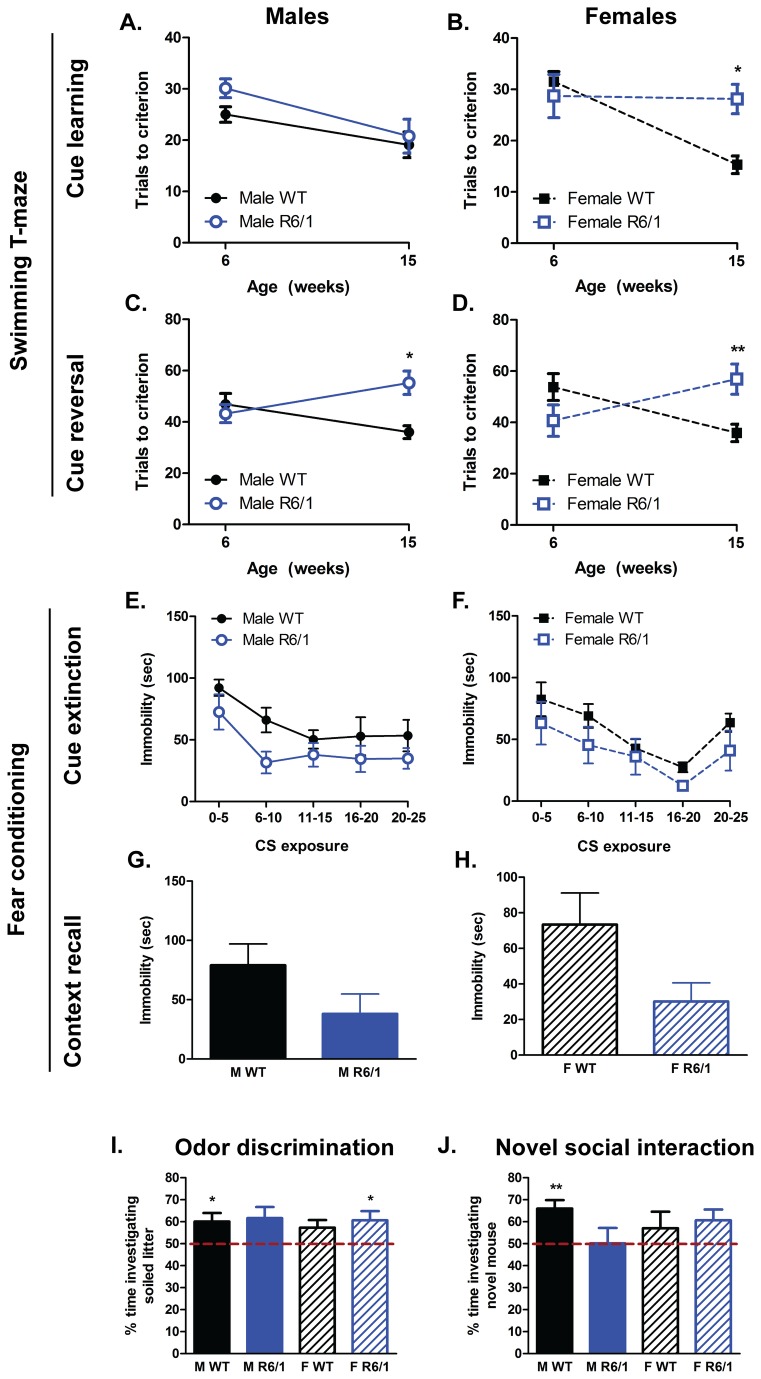
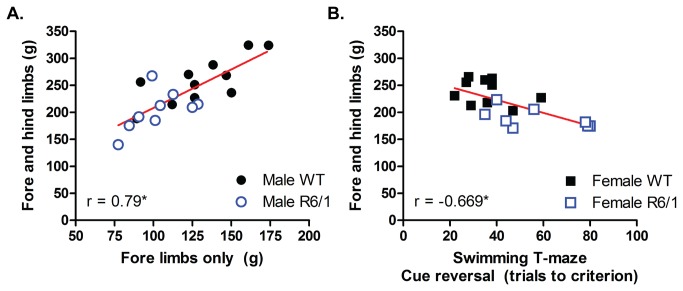
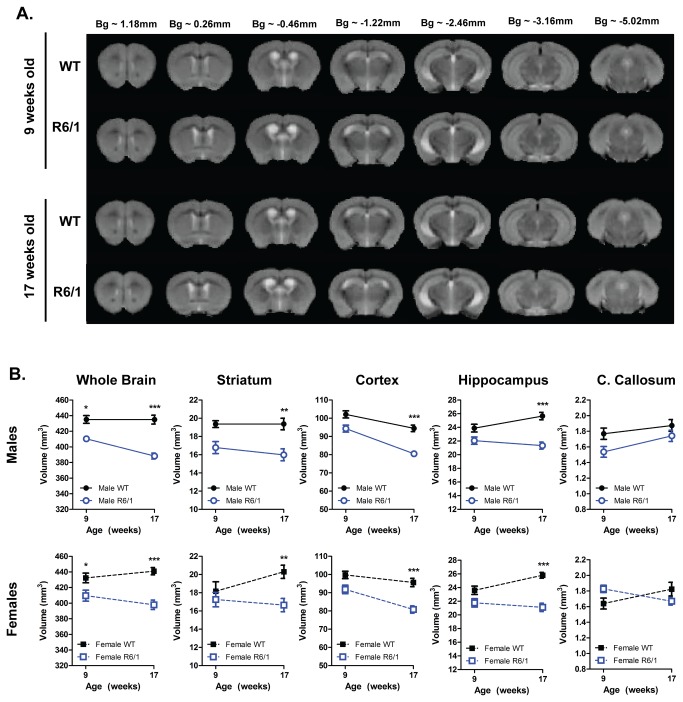
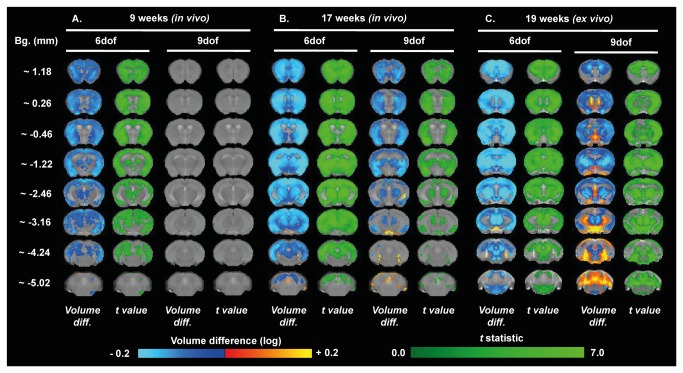
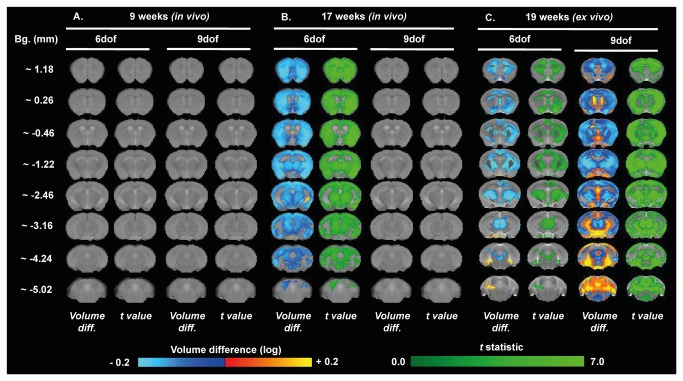
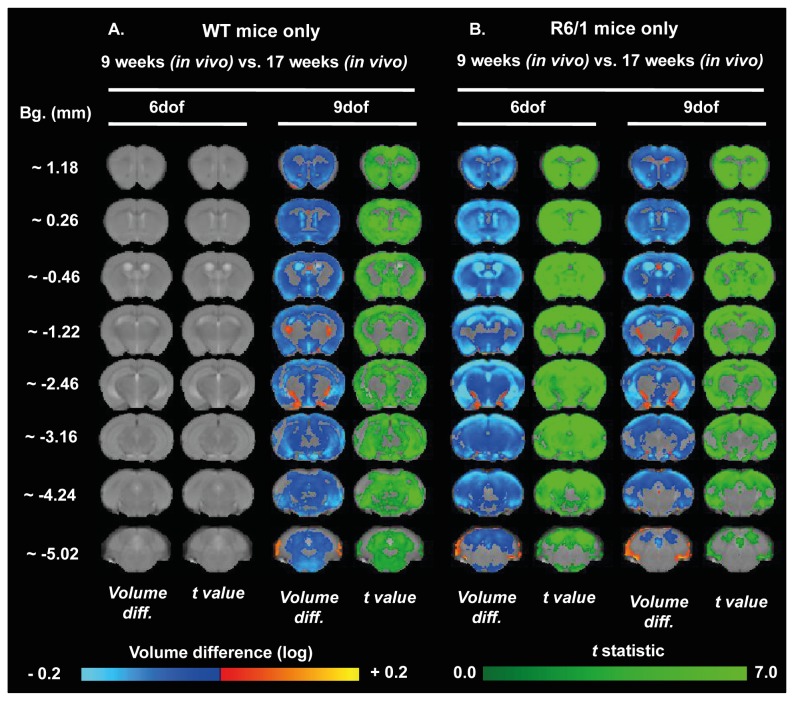
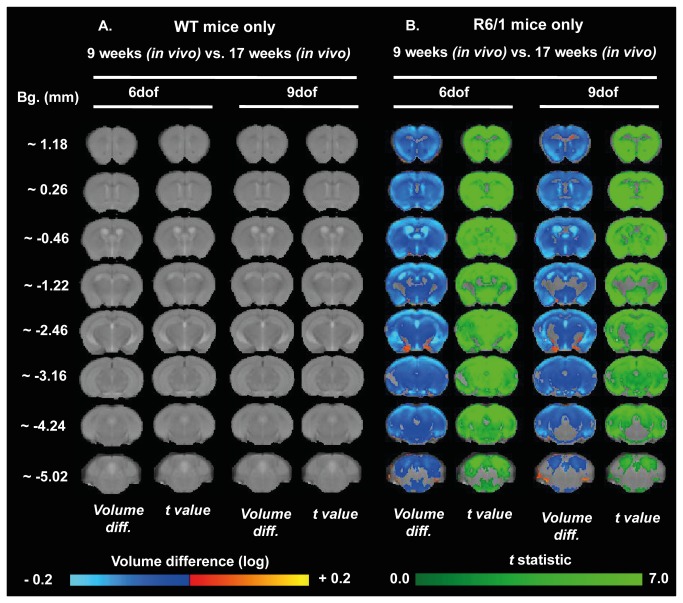
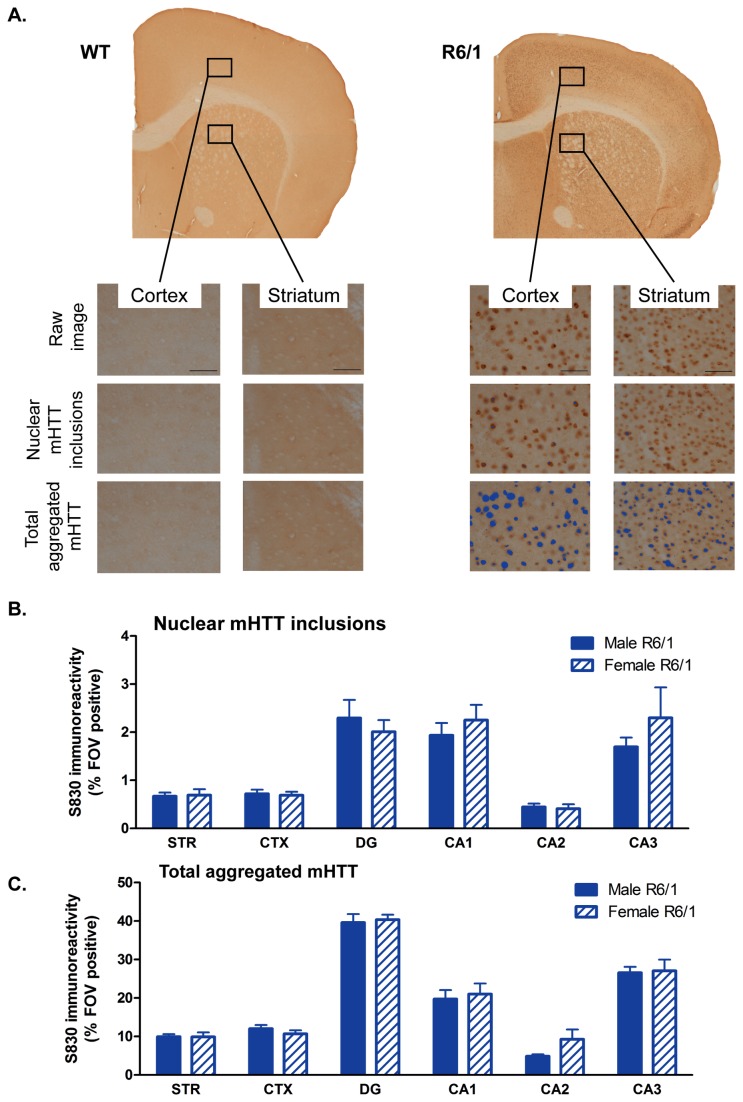
Huntington's disease (HD) is caused by the expansion of a CAG repeat in the huntingtin (HTT) gene. The R6 mouse models of HD express a mutant version of exon 1 HTT and typically develop motor and cognitive impairments, a widespread huntingtin (HTT) aggregate pathology and brain atrophy. Unlike the more commonly used R6/2 mouse line, R6/1 mice have fewer CAG repeats and, subsequently, a less rapid pathological decline. Compared to the R6/2 line, fewer descriptions of the progressive pathologies exhibited by R6/1 mice exist. The association between the molecular and cellular neuropathology with brain atrophy, and with the development of behavioral phenotypes remains poorly understood in many models of HD. In attempt to link these factors in the R6/1 mouse line, we have performed detailed assessments of behavior and of regional brain abnormalities determined through longitudinal, in vivo magnetic resonance imaging (MRI), as well as an end-stage, ex vivo MRI study and histological assessment. We found progressive decline in both motor and non-motor related behavioral tasks in R6/1 mice, first evident at 11 weeks of age. Regional brain volumes were generally unaffected at 9 weeks, but by 17 weeks there was significant grey matter atrophy. This age-related brain volume loss was validated using a more precise, semi-automated Tensor Based morphometry assessment. As well as these clear progressive phenotypes, mutant HTT (mHTT) protein, the hallmark of HD molecular pathology, was widely distributed throughout the R6/1 brain and was accompanied by neuronal loss. Despite these seemingly concomitant, robust pathological phenotypes, there appeared to be little correlation between the three main outcome measures: behavioral performance, MRI-detected brain atrophy and histopathology. In conclusion, R6/1 mice exhibit many features of HD, but the underlying mechanisms driving these clear behavioral disturbances and the brain volume loss, still remain unclear.
亨廷顿病(HD)是由亨廷顿(HTT)基因中 CAG 重复扩展引起的。R6 小鼠模型表达突变型 HTT 外显子 1,通常会出现运动和认知障碍、广泛的亨廷顿(HTT)聚集体病理学和脑萎缩。与更常用的 R6/2 小鼠系相比,R6/1 小鼠的 CAG 重复较少,因此病理下降速度较慢。与 R6/2 系相比,R6/1 小鼠表现出的进行性病理变化的描述较少。在许多 HD 模型中,分子和细胞神经病理学与脑萎缩以及行为表型的发展之间的联系仍知之甚少。为了在 R6/1 小鼠系中建立这些因素之间的联系,我们进行了详细的行为评估和通过纵向、体内磁共振成像(MRI)确定的区域脑异常评估,以及终末期、离体 MRI 研究和组织学评估。我们发现 R6/1 小鼠的运动和非运动相关行为任务都出现了进行性下降,最早在 11 周龄时出现。9 周时区域脑体积通常不受影响,但到 17 周时,灰质明显萎缩。使用更精确的半自动化张量形态计量学评估验证了这种与年龄相关的脑体积损失。除了这些明显的进行性表型外,突变 HTT(mHTT)蛋白是 HD 分子病理学的标志,广泛分布在 R6/1 大脑中,并伴有神经元丢失。尽管这些病理表型似乎同时存在,但行为表现、MRI 检测到的脑萎缩和组织病理学之间似乎没有明显的相关性。总之,R6/1 小鼠表现出许多 HD 特征,但驱动这些明显行为障碍和脑体积损失的潜在机制仍不清楚。