Mirabello Claudio, Adelfio Alessandro, Pollastri Gianluca
School of Computer Science and Informatics, University College Dublin, Belfield, Dublin 4, Ireland.
Biomolecules. 2014 Feb 10;4(1):160-80. doi: 10.3390/biom4010160.
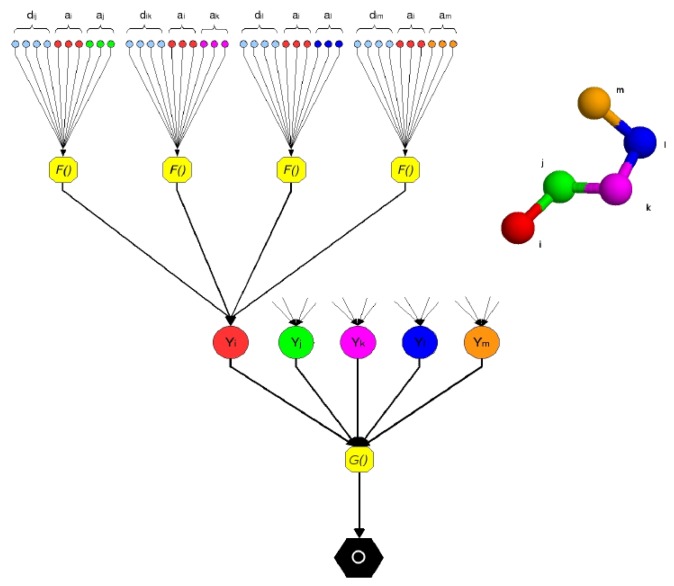
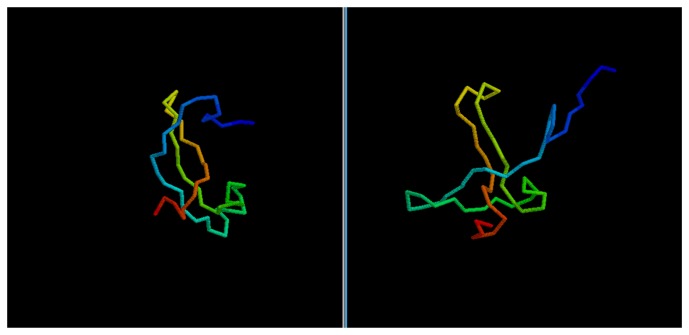
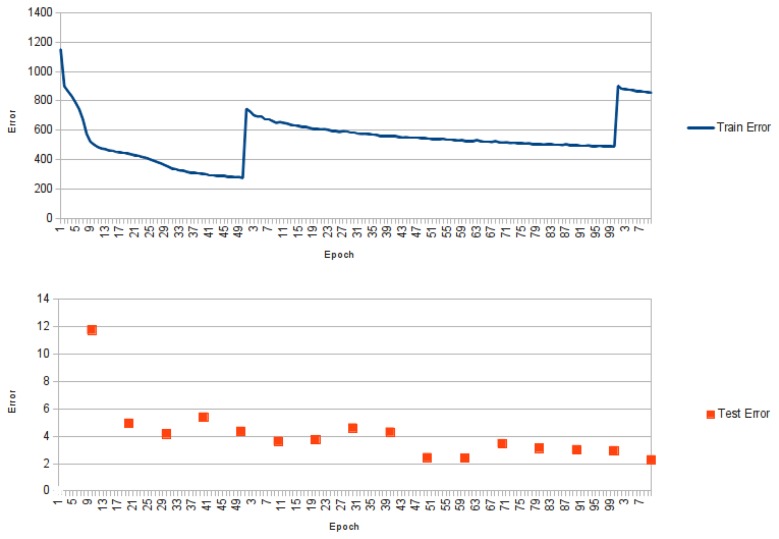
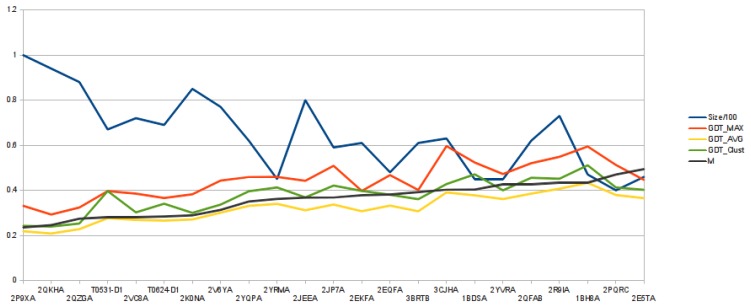
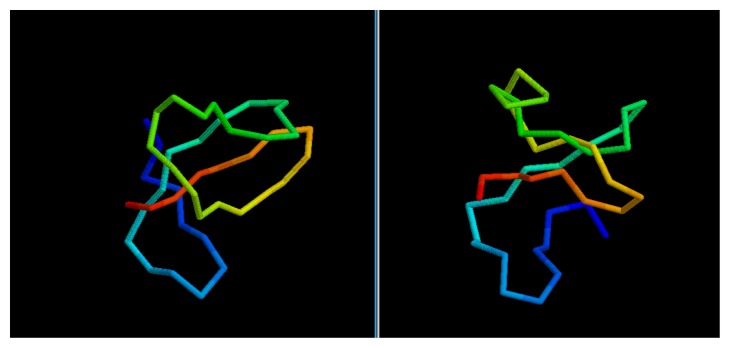
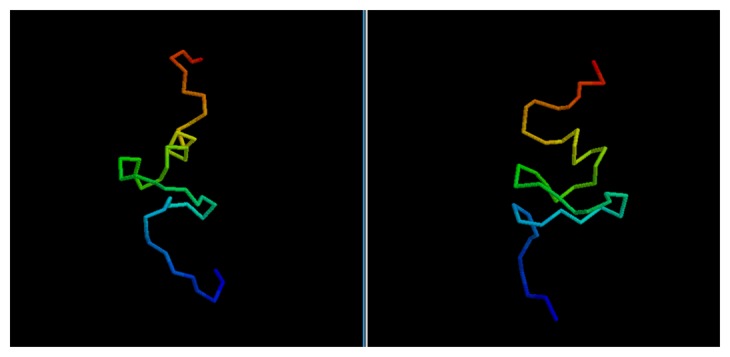
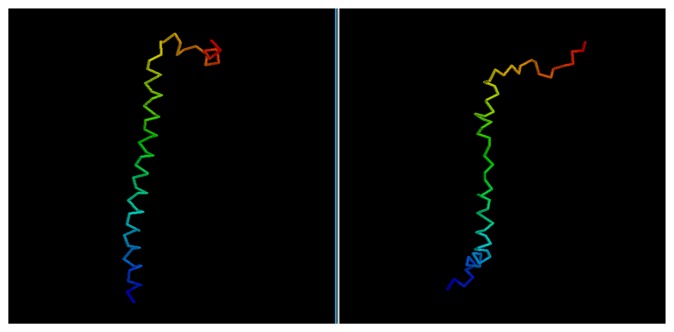
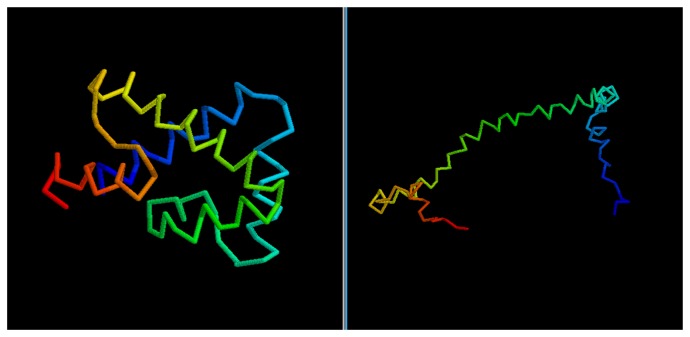
Predicting the fold of a protein from its amino acid sequence is one of the grand problems in computational biology. While there has been progress towards a solution, especially when a protein can be modelled based on one or more known structures (templates), in the absence of templates, even the best predictions are generally much less reliable. In this paper, we present an approach for predicting the three-dimensional structure of a protein from the sequence alone, when templates of known structure are not available. This approach relies on a simple reconstruction procedure guided by a novel knowledge-based evaluation function implemented as a class of artificial neural networks that we have designed: Neural Network Pairwise Interaction Fields (NNPIF). This evaluation function takes into account the contextual information for each residue and is trained to identify native-like conformations from non-native-like ones by using large sets of decoys as a training set. The training set is generated and then iteratively expanded during successive folding simulations. As NNPIF are fast at evaluating conformations, thousands of models can be processed in a short amount of time, and clustering techniques can be adopted for model selection. Although the results we present here are very preliminary, we consider them to be promising, with predictions being generated at state-of-the-art levels in some of the cases.
从氨基酸序列预测蛋白质的折叠是计算生物学中的重大问题之一。虽然在解决该问题方面已取得进展,特别是当蛋白质可以基于一个或多个已知结构(模板)进行建模时,但在没有模板的情况下,即使是最佳预测通常也远不可靠。在本文中,我们提出了一种在没有已知结构模板时仅从序列预测蛋白质三维结构的方法。这种方法依赖于一种简单的重建过程,该过程由我们设计的一类人工神经网络实现的基于新知识的评估函数引导:神经网络成对相互作用场(NNPIF)。该评估函数考虑每个残基的上下文信息,并通过使用大量诱饵作为训练集进行训练,以从非天然样构象中识别天然样构象。训练集在连续的折叠模拟过程中生成并迭代扩展。由于NNPIF在评估构象时速度很快,因此可以在短时间内处理数千个模型,并且可以采用聚类技术进行模型选择。尽管我们在此展示的结果非常初步,但我们认为它们很有前景,在某些情况下预测达到了当前的先进水平。