Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China.
Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China ; School of Life Science and Technology, ShanghaiTech University, Shanghai 200031, China.
J Cheminform. 2014 Jun 18;6:33. doi: 10.1186/1758-2946-6-33. eCollection 2014.
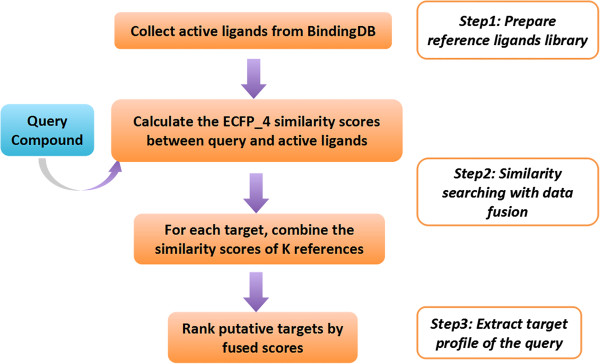
Ligand-based in silico target fishing can be used to identify the potential interacting target of bioactive ligands, which is useful for understanding the polypharmacology and safety profile of existing drugs. The underlying principle of the approach is that known bioactive ligands can be used as reference to predict the targets for a new compound.
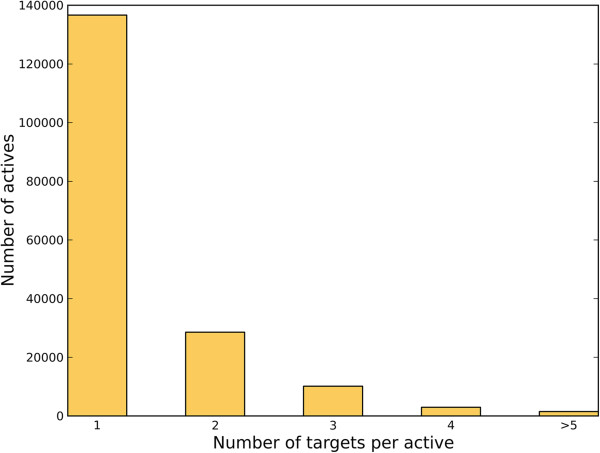
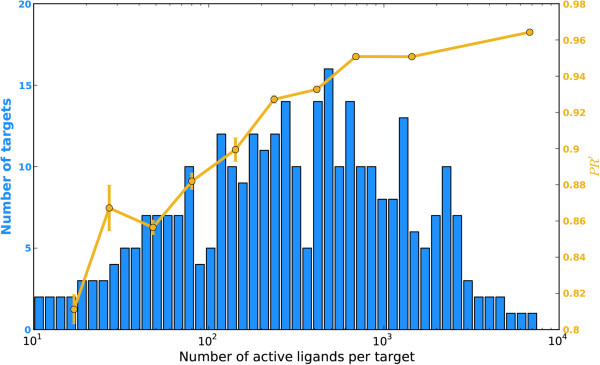
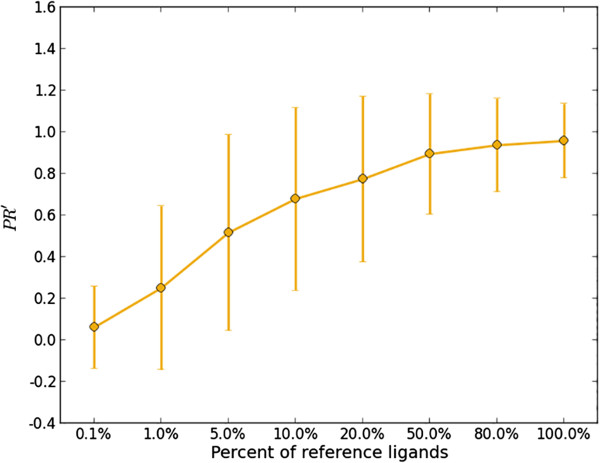
We tested a pipeline enabling large-scale target fishing and drug repositioning, based on simple fingerprint similarity rankings with data fusion. A large library containing 533 drug relevant targets with 179,807 active ligands was compiled, where each target was defined by its ligand set. For a given query molecule, its target profile is generated by similarity searching against the ligand sets assigned to each target, for which individual searches utilizing multiple reference structures are then fused into a single ranking list representing the potential target interaction profile of the query compound. The proposed approach was validated by 10-fold cross validation and two external tests using data from DrugBank and Therapeutic Target Database (TTD). The use of the approach was further demonstrated with some examples concerning the drug repositioning and drug side-effects prediction. The promising results suggest that the proposed method is useful for not only finding promiscuous drugs for their new usages, but also predicting some important toxic liabilities.
With the rapid increasing volume and diversity of data concerning drug related targets and their ligands, the simple ligand-based target fishing approach would play an important role in assisting future drug design and discovery.
基于配体的计算目标钓取可用于鉴定生物活性配体的潜在相互作用靶标,这对于理解现有药物的多药理学和安全性概况很有用。该方法的基本原理是可以将已知的生物活性配体用作参考,以预测新化合物的靶标。
我们测试了一种基于简单指纹相似性排名和数据融合的大规模目标钓取和药物重定位的管道。我们编译了一个包含 533 个药物相关靶标和 179807 个活性配体的大型库,其中每个靶标都由其配体集定义。对于给定的查询分子,通过与分配给每个靶标的配体集进行相似性搜索来生成其靶标谱,然后将针对多个参考结构的单独搜索融合到单个排名列表中,代表查询化合物的潜在靶标相互作用谱。该方法通过 10 折交叉验证和使用来自 DrugBank 和治疗靶标数据库(TTD)的数据的两个外部测试进行了验证。该方法还通过一些涉及药物重定位和药物副作用预测的示例进行了演示。有希望的结果表明,该方法不仅可用于寻找具有新用途的混杂药物,而且还可用于预测一些重要的毒性负担。
随着与药物相关靶标及其配体相关的数据量和多样性的快速增加,基于配体的简单目标钓取方法将在辅助未来药物设计和发现方面发挥重要作用。