Ohmura Itta, Morimoto Gentaro, Ohno Yousuke, Hasegawa Aki, Taiji Makoto
Laboratory for Computational Molecular Design, RIKEN QBiC (Quantitative Biology Center), 6F, 1-6-5, Minatojima-minamimachi, Chuo-ku, Kobe, Hyogo 650-0047, Japan.
Laboratory for Computational Molecular Design, RIKEN QBiC (Quantitative Biology Center), 6F, 1-6-5, Minatojima-minamimachi, Chuo-ku, Kobe, Hyogo 650-0047, Japan
Philos Trans A Math Phys Eng Sci. 2014 Aug 6;372(2021). doi: 10.1098/rsta.2013.0387.
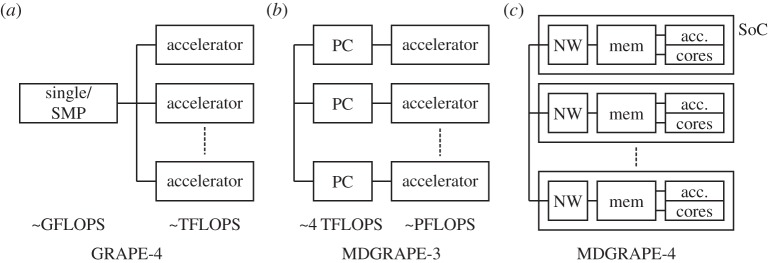
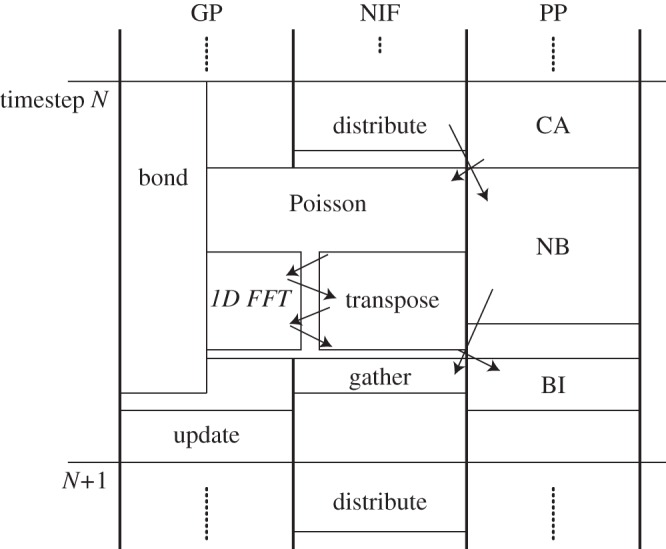
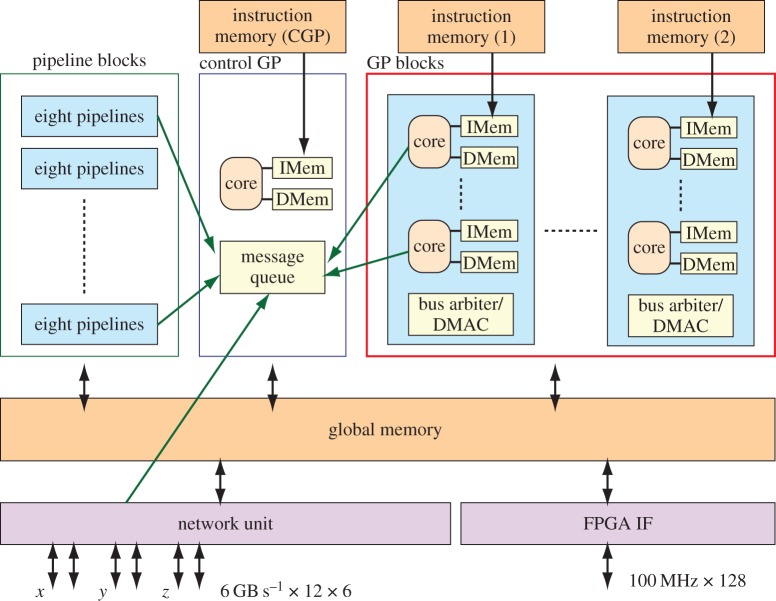
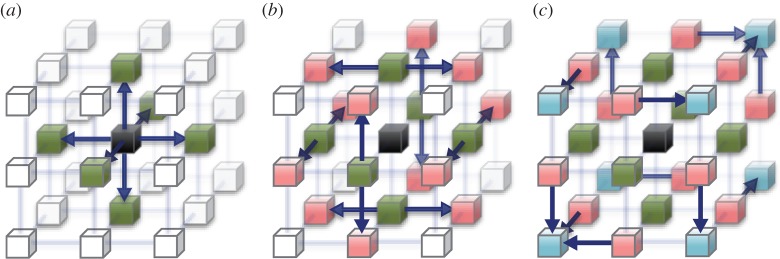
We are developing the MDGRAPE-4, a special-purpose computer system for molecular dynamics (MD) simulations. MDGRAPE-4 is designed to achieve strong scalability for protein MD simulations through the integration of general-purpose cores, dedicated pipelines, memory banks and network interfaces (NIFs) to create a system on chip (SoC). Each SoC has 64 dedicated pipelines that are used for non-bonded force calculations and run at 0.8 GHz. Additionally, it has 65 Tensilica Xtensa LX cores with single-precision floating-point units that are used for other calculations and run at 0.6 GHz. At peak performance levels, each SoC can evaluate 51.2 G interactions per second. It also has 1.8 MB of embedded shared memory banks and six network units with a peak bandwidth of 7.2 GB s(-1) for the three-dimensional torus network. The system consists of 512 (8×8×8) SoCs in total, which are mounted on 64 node modules with eight SoCs. The optical transmitters/receivers are used for internode communication. The expected maximum power consumption is 50 kW. While MDGRAPE-4 software has still been improved, we plan to run MD simulations on MDGRAPE-4 in 2014. The MDGRAPE-4 system will enable long-time molecular dynamics simulations of small systems. It is also useful for multiscale molecular simulations where the particle simulation parts often become bottlenecks.
我们正在研发MDGRAPE - 4,这是一种用于分子动力学(MD)模拟的专用计算机系统。MDGRAPE - 4旨在通过集成通用内核、专用流水线、内存库和网络接口(NIF)来创建片上系统(SoC),从而实现蛋白质MD模拟的强大可扩展性。每个SoC有64条专用流水线,用于非键合力计算,运行频率为0.8 GHz。此外,它还有65个带有单精度浮点单元的Tensilica Xtensa LX内核,用于其他计算,运行频率为0.6 GHz。在峰值性能水平下,每个SoC每秒可评估51.2 G次相互作用。它还具有1.8 MB的嵌入式共享内存库和六个网络单元,用于三维环形网络,峰值带宽为7.2 GB s⁻¹。该系统总共由512个(8×8×8)SoC组成,安装在64个带有八个SoC的节点模块上。光发射器/接收器用于节点间通信。预期最大功耗为50 kW。虽然MDGRAPE - 4软件仍在改进,但我们计划在2014年在MDGRAPE - 4上运行MD模拟。MDGRAPE - 4系统将能够对小系统进行长时间的分子动力学模拟。它对于多尺度分子模拟也很有用,在这种模拟中,粒子模拟部分常常成为瓶颈。